

mirpy#
ML-oriented embeddings for immune receptor repertoires (TCR/BCR) — the antigenomics group’s
machine-learning / embedding library (PyPI mirpy-lib, import mir).
Pure-Python and built on the antigenomics ecosystem (seqtree, vdjtools, arda).
mirpy turns receptor sequences into fixed-length vectors at three scales — the clonotype
(one receptor → a point), the repertoire (a whole sample → one fingerprint), and the cohort
(many donors → a comparable table) — and gives you the operations to measure across them
(distance, enrichment, MMD), read out what drives a signal, and decode embeddings back to
sequences. The classical v1.x/v2 toolkit is frozen on the legacy-v2 branch (mirpy-lib 2.x).
New here? Start with the user guide (install → a real result, one task per section),
skim the runnable examples, and reach for the API reference for
signatures. The mathematical theory (T1–T7) and recorded benchmark numbers live in the companion
2026-mirpy-analysis repo (benchmarks/{THEORY,BENCHMARKS}.md),
with the LaTeX theory appendix in 2026-mirpy-ms.
30-second quickstart#
import polars as pl
from mir.embedding.tcremp import TCREmp
model = TCREmp.from_defaults("human", "TRB", n_prototypes=1000)
df = pl.DataFrame({
"v_call": ["TRBV10-3*01", "TRBV20-1*01"],
"j_call": ["TRBJ2-7*01", "TRBJ1-2*01"],
"junction_aa": ["CASSIRSSYEQYF", "CSARVSGYYGYTF"],
})
X = model.embed(df) # (2, 3K) float32 — distances to 3K prototypes, per V/J/junction
Distance in the prototype-embedding space approximates the pairwise alignment distance (Theory T1);
TCREmp computes the junction part via seqtree.gapblock and adds baked germline V/J distances.
Or from the shell — pip install mirpy-lib ships a mir command:
mir embed clonotypes sample.tsv -o clonotypes.parquet # per-clonotype embedding table
mir embed repertoires cohort/*.tsv.gz -o phi.tsv --mmd mmd.tsv # one Φ(S) per sample, per chain
From a bag of receptors to one comparable fingerprint#
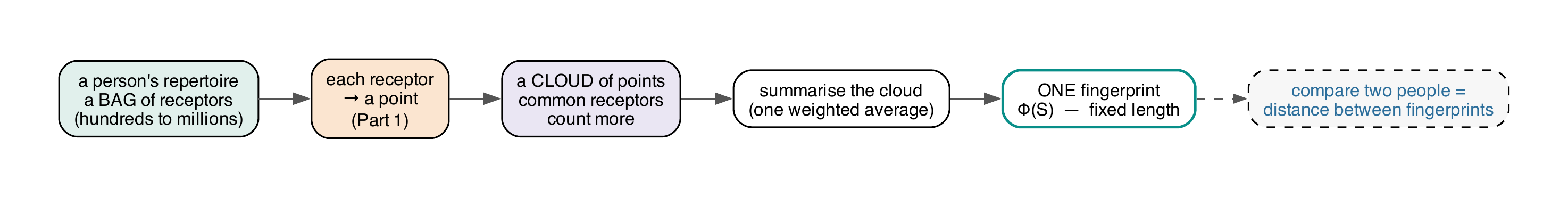
The sample-level embedding Φ(S) (mir.repertoire) sketches a repertoire as a weighted cloud
of clonotype points — depth-robust down into the RNA-seq regime (Theory T7):
from mir.repertoire import fit_repertoire_space, sample_embedding, mmd_matrix
space = fit_repertoire_space(model, pooled_clonotypes) # ONE basis for the cohort
embs = [sample_embedding(space, s) for s in samples] # Φ(S): mean ‖ diversity ‖ second moment
D = mmd_matrix(embs, unbiased=True) # pairwise repertoire distance (MMD²)
Capabilities (see the API reference)#
Clonotype embedding — TCREMP prototype embedding (
mir.embedding) onseqtree.gapblock+ baked arda germline distances (mir.distances), with PCA denoise and per-chain presets.Density — graph-free TCRNET/ALICE neighbour enrichment in embedding space, with an abundance channel and exact / kdtree / ANN backends (
mir.density, T6).Repertoire embedding — one fixed vector per sample: RFF kernel mean, coverage-standardised Hill diversity, and a second-moment Fisher block; MMD / HLA-stratified distance and a motif witness (
mir.repertoire, T7).Digital donor / cohort — fuse per-chain repertoire embeddings into one multi-chain donor matrix, hash-verified and serialisable, with batch residualisation and sample clustering (
mir.cohort, T7).Explainable readouts — name
Φ’s channels and ablate them under your scorer to see which one carries the signal, then hop from the winning kernel-mean channel to the clonotypes driving it (mir.explain+ scorers inmir.bench.eval, T7).Neural codecs — forward / inverse / Pgen / unified codecs and a learned repertoire set encoder; CUDA → MPS → CPU device selection (
mir.ml,[ml]extra).Sub-probability embeddings — a sample that was never fully observed carries a deficient mass instead of asserting full confidence: Good–Turing / Chao missing mass, a germline reference for the unseen, and the signed contrast
Ψ = mass·(Φ − naive)(mir.repertoire).Benchmark harness — VDJdb clustering + F1/retention and reproduced theory (
mir.bench).
Every formula, derivation and invariant behind these is collected in Mathematical foundations.