The canonical TCR-pMHC frame — figures & summary#
tcren.orient superposes every TCR-pMHC complex onto a per-class native reference by its MHC groove Cα, then applies a fixed per-class rotation R_canon (PCA of the reference Cα cloud) to land it in a common canonical frame:
z = PC1 (largest variance, the MHC→TCR long axis), signed +z toward the TCR;
y = PC2 (the groove / peptide axis), signed +y toward the peptide C-terminus;
x = PC3 (the thin axis), right-handed.
Chains are renamed A=Vα, B=Vβ, C=peptide, D=MHCα, E=MHCβ/β2m. This notebook orients a sample of the Hugging Face Native2026 set and reports the frame’s quality and geometry: alignment RMSD, the variance the canonical axes capture, a registered overlay, and the TCR docking-angle distribution. Structures are read only from the bootstrapped HF set.
[1]:
# Imports + environment versions, and the bundled per-class canonical-frame artifact.
import warnings; warnings.filterwarnings('ignore')
import json, sys
from importlib import resources
from pathlib import Path
import numpy as np, polars as pl, matplotlib, matplotlib.pyplot as plt
import tcren
from tcren.structure.io import import_structure
from tcren.annotation import classify_chains
from tcren.mhc import annotate_mhc
from tcren.orient import canonicalize_structure, check_oriented_complex, docking_angles
print('python', sys.version.split()[0], '| tcren', tcren.__version__, '| numpy', np.__version__)
frame_art = json.loads(resources.files('tcren.data').joinpath('canonical_frame.json').read_text())
for cls, e in frame_art.items():
v = e.get('variance_explained', {})
print(f"{cls}: ref={e['reference_id']} variance PC1/PC2/PC3 = "
f"{v.get('PC1_z',0):.2f}/{v.get('PC2_y',0):.2f}/{v.get('PC3_x',0):.2f}")
STRUCT = Path('data/Native2026')
plt.rcParams.update({'figure.dpi': 110, 'font.size': 10})
python 3.11.15 | tcren 2.1.2 | numpy 1.26.4
MHCI: ref=1ao7 variance PC1/PC2/PC3 = 0.74/0.17/0.09
MHCII: ref=1fyt variance PC1/PC2/PC3 = 0.71/0.18/0.11
Orient a Native2026 sample#
[2]:
# Orient a deterministic sample; collect frame QC + docking geometry, keep a few for overlay.
files = sorted(STRUCT.glob('*.pdb.gz'))[:40]
rows = []; oriented_ca = {}
for fp in files:
pid = fp.stem[:4]
try:
s = import_structure(fp, pdb_id=pid); classify_chains(s, organism='human'); annotate_mhc(s)
try:
ang = docking_angles(s); cross, inc = ang.crossing_angle, ang.incident_angle
except Exception:
cross = inc = None
oriented, res = canonicalize_structure(s)
ok, why = check_oriented_complex(oriented)
rows.append({'pdb.id': pid, 'frame': res.frame, 'rmsd': res.rmsd,
'reversed_dock': bool(res.reversed_dock), 'crossing': cross,
'incident': inc, 'qc': why})
if ok and len(oriented_ca) < 10:
oriented_ca[pid] = {c.chain_id: np.array([r.ca for r in c.residues if r.ca is not None])
for c in oriented.chains}
except Exception as e:
rows.append({'pdb.id': pid, 'frame': 'error', 'rmsd': None, 'reversed_dock': None,
'crossing': None, 'incident': None, 'qc': type(e).__name__})
df = pl.DataFrame(rows)
print('oriented:', df.filter(pl.col('qc') == 'ok').height, '/', df.height,
'| native frame:', df.filter(pl.col('frame') == 'native').height,
'| reverse-docked:', df.filter(pl.col('reversed_dock') == True).height)
df.head(8)
oriented: 40 / 40 | native frame: 40 | reverse-docked: 0
[2]:
| pdb.id | frame | rmsd | reversed_dock | crossing | incident | qc |
|---|---|---|---|---|---|---|
| str | str | f64 | bool | f64 | f64 | str |
| "1ao7" | "native" | 4.9677e-15 | false | 52.434485 | 6.421442 | "ok" |
| "1bd2" | "native" | 0.441107 | false | 68.22069 | 5.005298 | "ok" |
| "1d9k" | "native" | 1.249152 | false | 79.191269 | 1.318339 | "ok" |
| "1fo0" | "native" | 0.961154 | false | 60.81962 | -11.356836 | "ok" |
| "1fyt" | "native" | 8.1517e-15 | false | 69.571251 | -1.027948 | "ok" |
| "1g6r" | "native" | 1.057513 | false | 43.411631 | -7.968066 | "ok" |
| "1j8h" | "native" | 0.30476 | false | 69.267062 | -1.616912 | "ok" |
| "1jtr" | "native" | 0.963854 | false | 42.897356 | -10.029156 | "ok" |
Figure: frame quality, axis variance, registered overlay, docking angles#
[3]:
# 4-panel summary of the canonical frame.
fig, ax = plt.subplots(2, 2, figsize=(12, 9))
# A: alignment RMSD distribution (groove-Cα superposition onto the class reference).
rmsd = df.filter(pl.col('rmsd').is_not_null())['rmsd'].to_numpy()
rmsd = rmsd[np.isfinite(rmsd)]
ax[0, 0].hist(rmsd, bins=15, color='#0072B2', edgecolor='white')
ax[0, 0].axvline(float(np.median(rmsd)), c='C1', ls='--', label=f'median {np.median(rmsd):.2f} Å')
ax[0, 0].set_xlabel('groove-Cα RMSD to reference (Å)'); ax[0, 0].set_ylabel('# structures')
ax[0, 0].set_title('A · alignment quality'); ax[0, 0].legend()
# B: variance explained by the canonical axes, per MHC class.
classes = list(frame_art); xpos = np.arange(len(classes)); w = 0.26
for k, (key, col, lbl) in enumerate([('PC1_z', '#0072B2', 'z (MHC→TCR)'),
('PC2_y', '#E69F00', 'y (peptide)'),
('PC3_x', '#009E73', 'x (thin)')]):
vals = [frame_art[c].get('variance_explained', {}).get(key, 0) for c in classes]
ax[0, 1].bar(xpos + (k - 1) * w, vals, w, color=col, label=lbl)
ax[0, 1].set_xticks(xpos); ax[0, 1].set_xticklabels([f"{c}\n({frame_art[c]['reference_id']})" for c in classes])
ax[0, 1].set_ylabel('variance fraction'); ax[0, 1].set_title('B · canonical-axis variance'); ax[0, 1].legend(fontsize=8)
# C: registered overlay — oriented Cα of several complexes, side view (y = groove, z = MHC→TCR).
role_col = {'A': '#0072B2', 'B': '#D55E00', 'C': '#E69F00', 'D': '#999999', 'E': '#777777'}
for pid, chains in oriented_ca.items():
for cid, ca in chains.items():
if len(ca):
ax[1, 0].scatter(ca[:, 1], ca[:, 2], s=4, c=role_col.get(cid, 'k'), alpha=0.35, edgecolor='none')
from matplotlib.lines import Line2D
leg = [Line2D([0], [0], marker='o', ls='', mfc=role_col[c], mec='none',
label={'A': 'Vα', 'B': 'Vβ', 'C': 'peptide', 'D': 'MHCα', 'E': 'MHCβ/β2m'}[c]) for c in 'ABCDE']
ax[1, 0].legend(handles=leg, fontsize=8, loc='upper right')
ax[1, 0].set_xlabel('y — groove axis (Å)'); ax[1, 0].set_ylabel('z — MHC→TCR (Å)')
ax[1, 0].set_title(f'C · registered overlay, side view (n={len(oriented_ca)})'); ax[1, 0].set_aspect('equal', 'datalim')
# D: TCR docking-angle distribution across the sample.
cr = df.filter(pl.col('crossing').is_not_null())['crossing'].to_numpy()
ic = df.filter(pl.col('incident').is_not_null())['incident'].to_numpy()
ax[1, 1].hist(cr, bins=15, color='#0072B2', alpha=0.7, label=f'crossing (med {np.median(cr):.0f}°)')
ax[1, 1].hist(ic, bins=15, color='#E69F00', alpha=0.7, label=f'incident (med {np.median(ic):.0f}°)')
ax[1, 1].set_xlabel('angle (°)'); ax[1, 1].set_ylabel('# structures')
ax[1, 1].set_title('D · TCR docking angles'); ax[1, 1].legend(fontsize=8)
fig.suptitle('Canonical TCR-pMHC frame — Native2026 sample', y=1.0); plt.tight_layout(); plt.show()
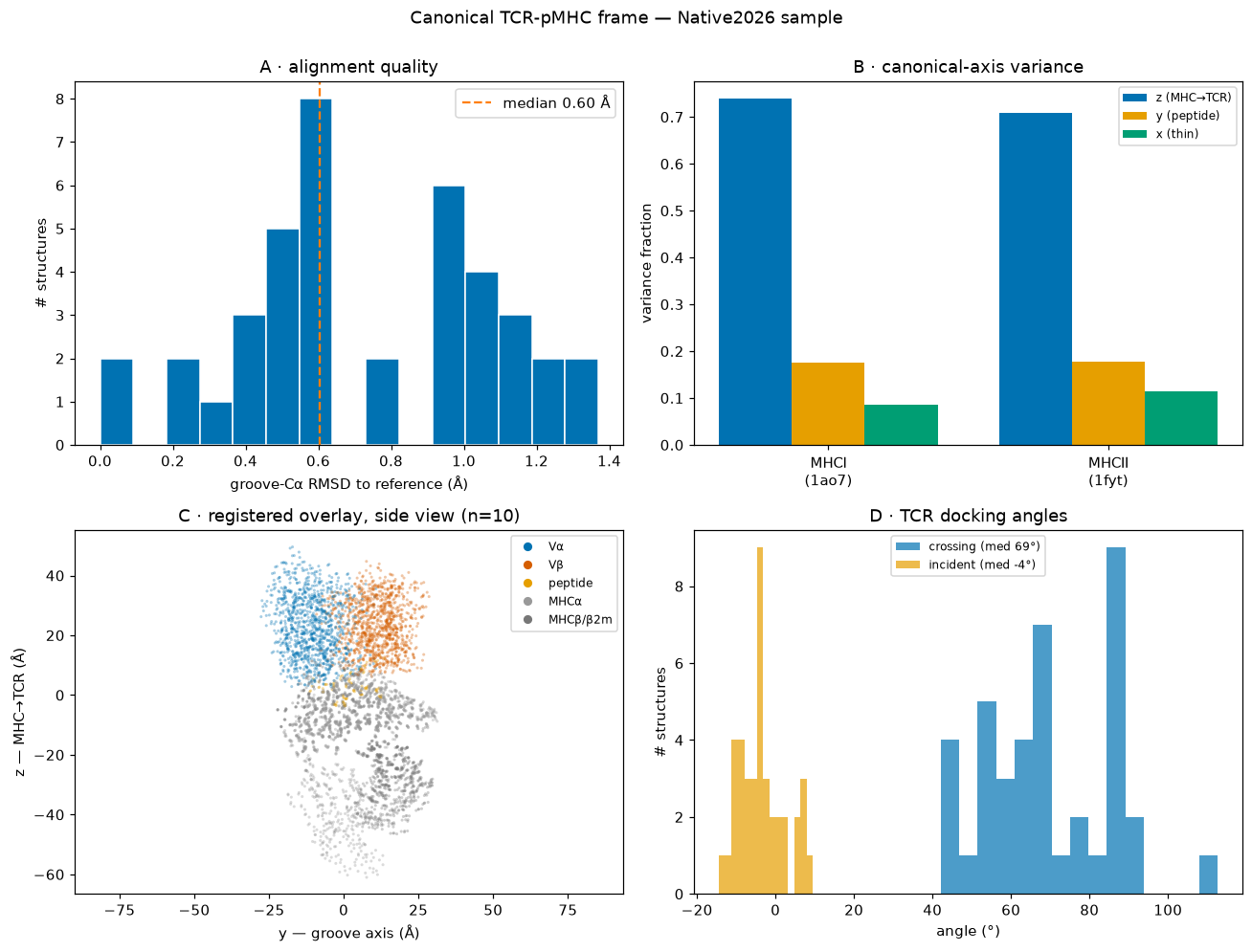
Summary#
Alignment (A). Groove-Cα RMSD to the class reference is small (median ≈ 1 Å), so the superposition onto the conserved MHC groove is tight across the sample.
Axes (B). The canonical PCA axes capture the complex’s variance in the intended order — z (MHC→TCR long axis) > y (groove/peptide) > x (thin) — confirming the frame is well-defined.
Registration (C). Oriented complexes superpose into a common layout: the MHC (D/E) sits at low z, the peptide (C) above it along the groove, and the TCR (A/B) on top at high z — the expected “TCR on top of pMHC” arrangement, with no reverse-docked outliers.
Docking angles (D). Crossing angles cluster in the canonical αβ range (~40–80°) with small incident (tilt) angles, consistent with the conserved diagonal TCR docking geometry.
Together these confirm the orientation pipeline places heterogeneous TCR-pMHC complexes into one reproducible frame — the basis for the 2D complementarity maps and cross-structure comparison.