2D complementarity maps#
Project the TCR-peptide-MHC interface (CDR1-3, peptide, MHC groove helices/floor) onto the canonical groove plane, build the two canonical polars tables (residue markup + contacts), and render a metadata-rich SVG complementarity map.
[1]:
# Environment versions (reproducibility)
import sys, polars as pl, tcren
print('python', sys.version.split()[0], '| polars', pl.__version__, '| tcren', tcren.__version__)
python 3.11.15 | polars 1.41.2 | tcren 2.1.2
[2]:
# Load a native complex, type chains (arda) and annotate the MHC groove
from tcren.structure import parse_structure
from tcren.annotation import classify_chains
from tcren.mhc import annotate_mhc
structure = parse_structure('data/Native2026/1ao7.pdb.gz', pdb_id='1ao7')
classify_chains(structure, organism='human')
annotate_mhc(structure)
[(c.chain_id, c.chain_type) for c in structure.chains]
[2]:
[('A', 'TRA'), ('B', 'TRB'), ('C', 'PEPTIDE'), ('D', 'MHCa'), ('E', 'B2M')]
[3]:
# Project onto the canonical groove plane and build the canonical tables
from tcren.project2d import (project_structure, residue_markup_table,
contacts_table, ca_contacts_table, pocket_markers)
projection = project_structure(structure)
markup = residue_markup_table(structure, projection)
contacts = contacts_table(structure, threshold=5.0)
ca_contacts = ca_contacts_table(structure, threshold=8.0)
pockets = pocket_markers(markup)
markup.head()
[3]:
shape: (5, 14)
| structure_id | structure_chain | complex_chain | complex_region | residue_index | aa_index | aa_len | aa | x | y | z | u | v | height |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| str | str | str | str | i64 | i64 | i64 | str | f64 | f64 | f64 | f64 | f64 | f64 |
| "1ao7" | "A" | "tra" | "fr1" | 1 | 0 | 110 | "K" | null | null | null | null | null | null |
| "1ao7" | "A" | "tra" | "fr1" | 2 | 1 | 110 | "E" | null | null | null | null | null | null |
| "1ao7" | "A" | "tra" | "fr1" | 3 | 2 | 110 | "V" | null | null | null | null | null | null |
| "1ao7" | "A" | "tra" | "fr1" | 4 | 3 | 110 | "E" | null | null | null | null | null | null |
| "1ao7" | "A" | "tra" | "fr1" | 5 | 4 | 110 | "Q" | null | null | null | null | null | null |
[4]:
# Contacts table is compatible with the original TCRen 5 A calculation
contacts.head()
[4]:
shape: (5, 11)
| structure_id | structure_chain_1 | structure_chain_2 | residue_index_1 | residue_index_2 | aa_index_1 | aa_index_2 | min_dist | contact_type | backbone_1 | backbone_2 |
|---|---|---|---|---|---|---|---|---|---|---|
| str | str | str | i64 | i64 | i64 | i64 | f64 | str | bool | bool |
| "1ao7" | "D" | "E" | 235 | 10 | 234 | 9 | 2.447153 | "hydrogen_bond" | true | false |
| "1ao7" | "A" | "C" | 31 | 5 | 30 | 4 | 2.54217 | "hydrogen_bond" | false | false |
| "1ao7" | "C" | "D" | 1 | 171 | 0 | 170 | 2.631378 | "hydrogen_bond" | true | false |
| "1ao7" | "A" | "B" | 50 | 105 | 49 | 100 | 2.63158 | "hydrogen_bond" | false | false |
| "1ao7" | "A" | "B" | 35 | 106 | 34 | 101 | 2.647912 | "hydrogen_bond" | false | true |
[5]:
# Render the SVG complementarity map (squares=residues, bold=Ca-Ca, dashed=inter-residue)
from tcren.viz import render_complementarity_map
from IPython.display import SVG
svg = render_complementarity_map(markup, contacts=contacts, ca_contacts=ca_contacts, pockets=pockets)
SVG(svg)
[5]:

Multiple views of the same complex#
The full map above shows the whole interface. Below are focused complementarity-map views (chain subsets — the TCR↔peptide footprint, and the peptide sitting in the MHC groove) followed by three orthogonal structural views of the oriented complex. Contacts in every view are the same closest-atom edges read off the contact table.
[6]:
# Different complementarity-map VIEWS via chain subsets (edges = contacts read off the map).
from IPython.display import display
map_views = {'TCR (CDR1-3) ↔ peptide': ['tra', 'trb', 'peptide'],
'peptide in the MHC groove': ['peptide', 'mhca', 'mhcb']}
for title, chains in map_views.items():
print(title)
display(SVG(render_complementarity_map(
markup, contacts=contacts, ca_contacts=ca_contacts, pockets=pockets, show_chains=chains)))
TCR (CDR1-3) ↔ peptide

peptide in the MHC groove

[7]:
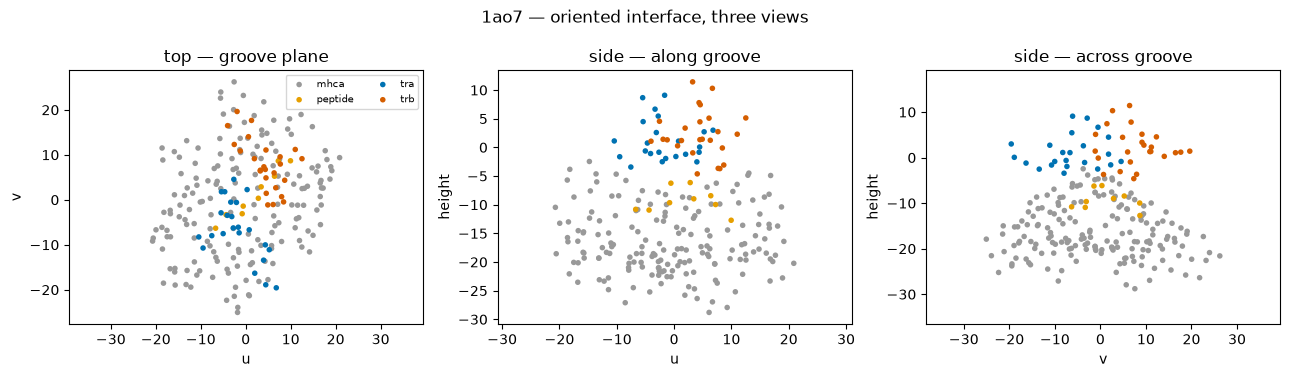
# Different STRUCTURAL views: the oriented interface in 3 orthogonal planes.
# u = along the groove (peptide N->C), v = across the groove, height = MHC->TCR normal.
# The canonical frame puts the TCR on top (+height) of the peptide/MHC.
import matplotlib.pyplot as plt
m = markup.filter(pl.col('u').is_not_null())
chain_col = {'mhca': '#999999', 'mhcb': '#777777', 'peptide': '#E69F00',
'tra': '#0072B2', 'trb': '#D55E00'}
planes = [('u', 'v', 'top — groove plane'),
('u', 'height', 'side — along groove'),
('v', 'height', 'side — across groove')]
fig, axes = plt.subplots(1, 3, figsize=(13, 3.8))
for ax, (a1, a2, ttl) in zip(axes, planes):
for ch, col in chain_col.items():
sub = m.filter(pl.col('complex_chain') == ch)
if sub.height:
ax.scatter(sub[a1], sub[a2], s=16, c=col, label=ch, edgecolor='none')
ax.set_xlabel(a1); ax.set_ylabel(a2); ax.set_title(ttl); ax.set_aspect('equal', 'datalim')
axes[0].legend(fontsize=7, ncol=2, loc='best')
fig.suptitle('1ao7 — oriented interface, three views'); plt.tight_layout(); plt.show()
[8]:
# Polars summarize: contact-type breakdown of the TCR (CDR) -> peptide interface
tcr_pep = (contacts
.join(markup.select('structure_chain', 'aa_index', 'complex_chain', 'complex_region'),
left_on=['structure_chain_1', 'aa_index_1'],
right_on=['structure_chain', 'aa_index'])
.filter(pl.col('complex_chain').is_in(['tra', 'trb'])
& pl.col('structure_chain_2').is_in([c.chain_id for c in structure.chains if c.chain_type=='PEPTIDE'])))
tcr_pep.group_by('complex_region', 'contact_type').agg(
pl.len().alias('n'), pl.col('min_dist').min().round(2).alias('closest')
).sort('complex_region', 'n', descending=[False, True])
[8]:
shape: (6, 4)
| complex_region | contact_type | n | closest |
|---|---|---|---|
| str | str | u32 | f64 |
| "cdr1" | "hydrogen_bond" | 4 | 2.54 |
| "cdr1" | "polar" | 3 | 3.66 |
| "cdr1" | "hydrophobic" | 1 | 4.93 |
| "cdr3" | "polar" | 13 | 3.32 |
| "cdr3" | "hydrogen_bond" | 4 | 2.89 |
| "cdr3" | "hydrophobic" | 4 | 3.66 |