Example — GILGFVFTL / HLA-A*02:01 and the CDR3β Arg–Ser motif#
The influenza M1₅₈₋₆₆ epitope GILGFVFTL presented by **HLA-A*02:01 is recognised by a strongly biased, public T-cell response built on TRBV19, whose hallmark is a conservedArg–Ser (RS) motif in CDR3β. The canonical structure is the JM22 TCR (PDB 1oga**), with CDR3β ASSSRSSYEQY — the Arg and the following Ser reach into the peptide.
This public RS motif is documented in VDJdb (Shugay M, Bagaev DV, Zvyagin IV, et al., Nucleic Acids Research 2018;46(D1):D419–D427, https://doi.org/10.1093/nar/gkx760; record retrieved from PubMed, PMID 28977646). Here we orient 1oga into the canonical frame and use the tcren complementarity maps to show where the Arg–Ser residues contact the peptide. The structure is read only from the bootstrapped HF Native2026 set.
[1]:
# Load the JM22 complex (1oga) from the HF set, type chains (arda), annotate the MHC groove.
import warnings; warnings.filterwarnings('ignore')
import sys
from pathlib import Path
import numpy as np, polars as pl, matplotlib, matplotlib.pyplot as plt
import tcren
from tcren.structure.io import import_structure
from tcren.annotation import classify_chains
from tcren.mhc import annotate_mhc
from tcren.orient import docking_angles
from tcren.project2d import (project_structure, residue_markup_table, contacts_table,
ca_contacts_table, pocket_markers, region_pair_contacts)
from tcren.viz import render_complementarity_map
from IPython.display import SVG, display
print('python', sys.version.split()[0], '| tcren', tcren.__version__)
s = import_structure(Path('data/Native2026/1oga.pdb.gz'), pdb_id='1oga')
classify_chains(s, organism='human'); annotate_mhc(s)
cdr3b = next(r.sequence for c in s.chains if c.chain_type == 'TRB'
for r in c.regions if r.region_type == 'CDR3')
pep = next(c.sequence() for c in s.chains if c.chain_type == 'PEPTIDE')
ang = docking_angles(s)
print(f'peptide={pep} CDR3β={cdr3b} crossing={ang.crossing_angle:.0f}° incident={ang.incident_angle:.0f}°')
python 3.11.15 | tcren 2.1.2
peptide=GILGFVFTL CDR3β=ASSSRSSYEQY crossing=83° incident=-14°
Where does the Arg–Ser motif touch the peptide?#
[2]:
# CDR3β → peptide contacts, annotated with the CDR3β residue identity; flag the Arg/Ser motif.
markup = residue_markup_table(s)
# aa per (complex_chain, region, aa_index) lookup for labelling contact ends.
aa_at = {(r['complex_chain'], r['complex_region'], r['aa_index']): (r['aa'], r['residue_index'])
for r in markup.iter_rows(named=True)}
rp = region_pair_contacts(s, kind='closest')
cdr3b_pep = rp.filter(
(((pl.col('complex_chain_1') == 'trb') & (pl.col('region_1') == 'cdr3') & (pl.col('complex_chain_2') == 'peptide')) |
((pl.col('complex_chain_2') == 'trb') & (pl.col('region_2') == 'cdr3') & (pl.col('complex_chain_1') == 'peptide'))))
rows = []
for r in cdr3b_pep.iter_rows(named=True):
# orient so end 1 = CDR3β, end 2 = peptide
if r['complex_chain_1'] == 'trb':
tcr_aa, tcr_resi = aa_at.get(('trb', 'cdr3', r['aa_index_1']), ('?', None))
pep_aa, pep_resi = aa_at.get(('peptide', 'peptide', r['aa_index_2']), ('?', None))
else:
tcr_aa, tcr_resi = aa_at.get(('trb', 'cdr3', r['aa_index_2']), ('?', None))
pep_aa, pep_resi = aa_at.get(('peptide', 'peptide', r['aa_index_1']), ('?', None))
rows.append({'cdr3b_res': f'{tcr_aa}{tcr_resi}', 'peptide_res': f'{pep_aa}{pep_resi}',
'min_dist': round(r['min_dist'], 2), 'contact_type': r['contact_type'],
'motif': tcr_aa in ('R', 'S')})
cdr3b_tbl = pl.DataFrame(rows).sort('min_dist')
print('CDR3β residues contacting the peptide:',
sorted({r['cdr3b_res'] for r in rows}))
cdr3b_tbl
CDR3β residues contacting the peptide: ['R98', 'S100', 'S99']
[2]:
| cdr3b_res | peptide_res | min_dist | contact_type | motif |
|---|---|---|---|---|
| str | str | f64 | str | bool |
| "S99" | "V6" | 3.09 | "hydrogen_bond" | true |
| "R98" | "F5" | 3.78 | "polar" | true |
| "S100" | "F5" | 3.82 | "polar" | true |
| "S99" | "F5" | 4.03 | "polar" | true |
| "R98" | "F7" | 4.16 | "hydrophobic" | true |
| "R98" | "V6" | 4.84 | "polar" | true |
Complementarity map and multiple structural views#
[3]:
# Project onto the canonical groove plane and build the canonical tables.
proj = project_structure(s)
markup = residue_markup_table(s, proj)
contacts = contacts_table(s, threshold=5.0)
ca_contacts = ca_contacts_table(s, threshold=8.0)
pockets = pocket_markers(markup)
# Full map, then the focused TCR(CDR1-3)↔peptide footprint.
print('full interface'); display(SVG(render_complementarity_map(markup, contacts=contacts, ca_contacts=ca_contacts, pockets=pockets)))
print('TCR (CDR1-3) ↔ peptide'); display(SVG(render_complementarity_map(
markup, contacts=contacts, ca_contacts=ca_contacts, pockets=pockets, show_chains=['tra', 'trb', 'peptide'])))
full interface

TCR (CDR1-3) ↔ peptide

[4]:
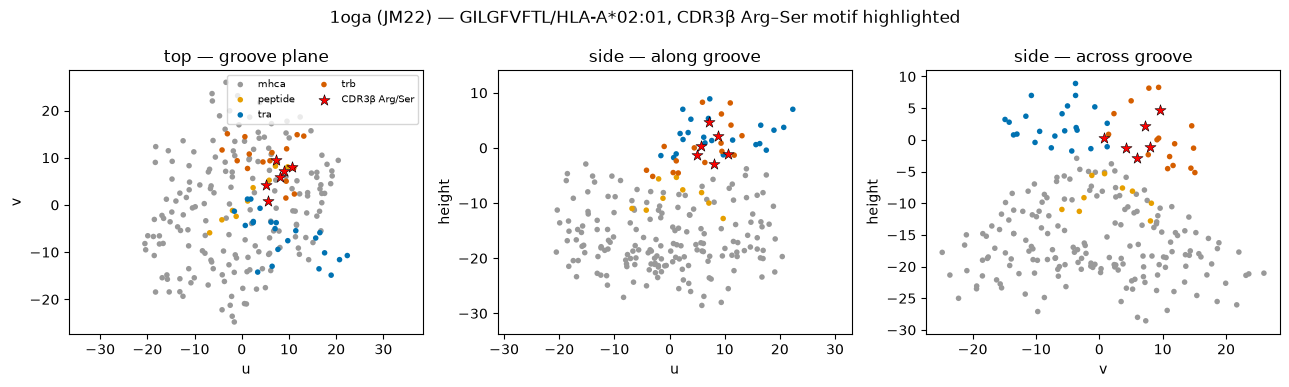
# Three orthogonal structural views; the CDR3β Arg/Ser residues highlighted in red.
m = markup.filter(pl.col('u').is_not_null())
chain_col = {'mhca': '#999999', 'mhcb': '#777777', 'peptide': '#E69F00', 'tra': '#0072B2', 'trb': '#D55E00'}
is_rs = (pl.col('complex_chain') == 'trb') & (pl.col('complex_region') == 'cdr3') & (pl.col('aa').is_in(['R', 'S']))
planes = [('u', 'v', 'top — groove plane'), ('u', 'height', 'side — along groove'), ('v', 'height', 'side — across groove')]
fig, axes = plt.subplots(1, 3, figsize=(13, 3.9))
for ax, (a1, a2, ttl) in zip(axes, planes):
for ch, col in chain_col.items():
sub = m.filter((pl.col('complex_chain') == ch) & ~is_rs)
if sub.height:
ax.scatter(sub[a1], sub[a2], s=16, c=col, label=ch, edgecolor='none')
rs = m.filter(is_rs)
if rs.height:
ax.scatter(rs[a1], rs[a2], s=70, marker='*', c='red', label='CDR3β Arg/Ser', edgecolor='k', linewidth=0.4, zorder=5)
ax.set_xlabel(a1); ax.set_ylabel(a2); ax.set_title(ttl); ax.set_aspect('equal', 'datalim')
axes[0].legend(fontsize=7, ncol=2, loc='best')
fig.suptitle('1oga (JM22) — GILGFVFTL/HLA-A*02:01, CDR3β Arg–Ser motif highlighted'); plt.tight_layout(); plt.show()
Summary#
Oriented into the canonical frame, the JM22 TCR docks GILGFVFTL/HLA-A02:01 at a crossing angle in the canonical αβ range. The complementarity map and the CDR3β→peptide contact table show the conserved Arg–Ser motif of CDR3β reaching across the peptide — the structural correlate of the public TRBV19 RS signature catalogued in VDJdb (Shugay et al., Nucleic Acids Research* 2018, https://doi.org/10.1093/nar/gkx760; via PubMed). The same recipe — import_structure →
classify_chains → annotate_mhc → project_structure → region_pair_contacts / render_complementarity_map — works for any TCR-pMHC complex in the HF set.