TCR Repertoire Aging — Functional Diversity Analysis#
Extends the basic aging analysis (aging_analysis.ipynb) with metaclonotype-based functional diversity metrics computed on 100 k-read-downsampled TRB repertoires from the Hugging Face AIRR benchmark (Britanova et al. 2016 aging cohort).
Metaclonotype clustering: 1-mismatch Hamming edit-distance graph, Louvain communities, minimum cluster size 2.
Metrics compared side-by-side:
Metric |
Clonotypic |
Functional |
|---|---|---|
Diversity (Shannon q=1) |
Shannon over clonotypes |
Shannon over metaclonotype clusters |
Rarefaction |
Clonotype-level iNEXT |
Metaclonotype-level rarefaction |
Pairwise F overlap |
Clonotypic F similarity (exact) |
Functional F (metaclonotype identity sharing) |
Vs-pool F overlap |
Clonotypic donor-vs-pool |
Functional donor-vs-pool |
Reference: Britanova OV et al. J Immunol. 2016.
[1]:
# Cell 1: Environment versions
import platform, importlib, sys
print('Python:', platform.python_version())
for pkg in ('numpy', 'polars', 'scipy', 'matplotlib', 'igraph', 'mir'):
try:
m = importlib.import_module(pkg)
print(f' {pkg}: {getattr(m, "__version__", "?")}')
except ImportError:
print(f' {pkg}: NOT INSTALLED')
Python: 3.12.12
numpy: 1.26.4
polars: 1.40.1
scipy: 1.17.1
matplotlib: 3.10.9
igraph: 1.0.0
mir: ?
[2]:
# Cell 2: Imports
import os
import time
import warnings
from pathlib import Path
import numpy as np
import polars as pl
from scipy import stats
import matplotlib.pyplot as plt
import matplotlib.ticker as mticker
from mir.common.parser import VDJtoolsParser
from mir.common.repertoire import LocusRepertoire
from mir.common.sampling import downsample
from mir.common.diversity import (
summarize_clonotypes,
hill_curve_clonotypes,
rarefaction_curve_clonotypes,
)
from mir.common.metaclonotype import (
functional_diversity,
functional_hill_curve,
functional_rarefaction_curve,
functional_overlap_1,
)
from mir.comparative.overlap import pairwise_overlap_matrix, many_vs_pool_overlap
from mir.comparative import clear_pairwise_target_cache
from mir.graph.edit_distance_graph import build_edit_distance_graph
from mir.utils.metaclonotype_clustering import metaclonotypes_from_graph_communities
from mir.utils.notebook_assets import ensure_airr_benchmark, find_repo_root
warnings.filterwarnings('ignore', category=FutureWarning)
warnings.filterwarnings('ignore', category=RuntimeWarning)
warnings.filterwarnings('ignore', category=UserWarning, module='mir.common.sampling')
plt.rcParams.update({
'figure.dpi': 150,
'font.family': 'STIXGeneral',
'axes.titlesize': 12,
'axes.labelsize': 11,
'xtick.labelsize': 10,
'ytick.labelsize': 10,
'legend.fontsize': 10,
'axes.spines.top': False,
'axes.spines.right': False,
'axes.linewidth': 0.8,
'grid.linewidth': 0.4,
})
DOWNSAMPLE_TARGET = 100_000
METACLONOTYPE_METHOD = 'louvain'
METACLONOTYPE_THRESHOLD = 1
METACLONOTYPE_MIN_SIZE = 2
N_JOBS = 4
clear_pairwise_target_cache()
EXECUTION_TIMES: dict[str, float] = {}
print('Setup complete.')
/Users/mikesh/vcs/mirpy/.venv/lib/python3.12/site-packages/tqdm/auto.py:21: TqdmWarning: IProgress not found. Please update jupyter and ipywidgets. See https://ipywidgets.readthedocs.io/en/stable/user_install.html
from .autonotebook import tqdm as notebook_tqdm
Setup complete.
[3]:
# Cell 3: Resolve data root and refresh AIRR benchmark
repo_root = find_repo_root(Path.cwd())
t0 = time.perf_counter()
benchmark_root = ensure_airr_benchmark(repo_root, allow_patterns=['vdjtools/**'])
EXECUTION_TIMES['asset_refresh_s'] = time.perf_counter() - t0
VDJTOOLS_DIR = benchmark_root / 'vdjtools'
META_PATH = VDJTOOLS_DIR / 'metadata_aging.txt'
if not META_PATH.exists():
raise FileNotFoundError(f'Missing {META_PATH}')
repertoire_lookup = {p.name: p for p in sorted(VDJTOOLS_DIR.glob('*.txt.gz'))}
print(f'Benchmark root : {benchmark_root}')
print(f'Indexed repertoires : {len(repertoire_lookup):,}')
Benchmark root : /Users/mikesh/vcs/mirpy/notebooks/assets/large/airr_benchmark
Indexed repertoires : 316
[4]:
# Cell 4: Load aging metadata
meta = pl.read_csv(META_PATH, separator='\t')
rename_map = {c: c.lstrip('#') for c in meta.columns if c.startswith('#')}
if rename_map:
meta = meta.rename(rename_map)
meta = meta.with_columns([
pl.col('sample_id').cast(pl.Utf8),
pl.col('file_name').cast(pl.Utf8),
pl.col('age').cast(pl.Int64),
])
print(f'Metadata rows : {meta.height} '
f'Age range: {meta["age"].min()}\u2013{meta["age"].max()} y '
f'Cord-blood: {meta.filter(pl.col("age")==0).height}')
meta.head(6).to_pandas()
Metadata rows : 79 Age range: 0–103 y Cord-blood: 8
[4]:
| file_name | sample_id | sex | age | label | |
|---|---|---|---|---|---|
| 0 | A3-i101.txt | A3-i101 | F | 36 | p1-F36 |
| 1 | A3-i102.txt | A3-i102 | F | 43 | p2-F43 |
| 2 | A3-i106.txt | A3-i106 | F | 43 | p3-F43 |
| 3 | A3-i107.txt | A3-i107 | F | 39 | p4-F39 |
| 4 | A3-i110.txt | A3-i110 | F | 34 | p5-F34 |
| 5 | A3-i145.txt | A3-i145 | F | 86 | p6-F86 |
[5]:
# Cell 5: Load and downsample repertoires to 100k reads
parser = VDJtoolsParser(sep='\t')
def _resolve(file_name: str) -> Path | None:
for cand in [file_name, f'{file_name}.gz', file_name.replace('.txt', '.txt.gz'), file_name.replace('.gz', '')]:
p = repertoire_lookup.get(cand)
if p is not None and p.exists():
return p
return None
reps, loaded_rows = [], []
t0 = time.perf_counter()
for row in meta.iter_rows(named=True):
p = _resolve(str(row['file_name']))
if p is None:
continue
clones = parser.parse(str(p))
rep = LocusRepertoire(clonotypes=clones, locus='TRB')
rep = downsample(rep, DOWNSAMPLE_TARGET, random_seed=42)
reps.append(rep)
loaded_rows.append({'sample_id': row['sample_id'], 'age': int(row['age']), 'sex': row.get('sex')})
EXECUTION_TIMES['load_s'] = time.perf_counter() - t0
meta_valid = pl.DataFrame(loaded_rows)
sample_ids = meta_valid['sample_id'].to_list()
ages = meta_valid['age'].to_list()
ages_arr = np.array(ages, dtype=float)
clonotype_counts = np.array([r.clonotype_count for r in reps], dtype=int)
print(f'Loaded {len(reps)} repertoires in {EXECUTION_TIMES["load_s"]:.1f}s')
print(f'Clonotype count: min={clonotype_counts.min():,} '
f'median={int(np.median(clonotype_counts)):,} '
f'max={clonotype_counts.max():,}')
Loaded 79 repertoires in 108.4s
Clonotype count: min=12,439 median=51,303 max=93,154
1 · Clonotypic diversity#
[6]:
# Cell 6: Clonotypic diversity per sample
div_rows = []
for sid, age, rep in zip(sample_ids, ages, reps):
d = summarize_clonotypes(rep.clonotypes)
div_rows.append({
'sample_id': sid, 'age': age,
'n_clonotypes': d.abundance,
'shannon': d.shannon,
'gini_simpson': d.gini_simpson,
'chao1': d.chao1,
})
clono_div_df = pl.DataFrame(div_rows)
fig, axes = plt.subplots(1, 3, figsize=(13, 4.2), constrained_layout=True)
for ax, (col, label) in zip(axes, [
('n_clonotypes', 'Observed clonotypes'),
('shannon', 'Shannon entropy (q=1)'),
('gini_simpson', 'Gini-Simpson index'),
]):
x = ages_arr
y = clono_div_df[col].to_numpy().astype(float)
ax.scatter(x, y, c=x, cmap='viridis', s=45, alpha=0.85, edgecolors='white', linewidths=0.25)
slope, intercept, r, p, _ = stats.linregress(x, y)
xr = np.linspace(x.min(), x.max(), 200)
ax.plot(xr, slope * xr + intercept, color='#C1121F', lw=2)
ax.set_xlabel('Age (years)')
ax.set_ylabel(label)
ax.set_title(label)
ax.text(0.03, 0.97, f'$r={r:.3f}$\n$p={p:.2e}$',
transform=ax.transAxes, va='top', fontsize=9,
bbox=dict(facecolor='white', alpha=0.75, edgecolor='none', pad=2))
fig.suptitle('Clonotypic Diversity vs Age (100k-read downsampled)', fontsize=13, y=1.02)
plt.show()
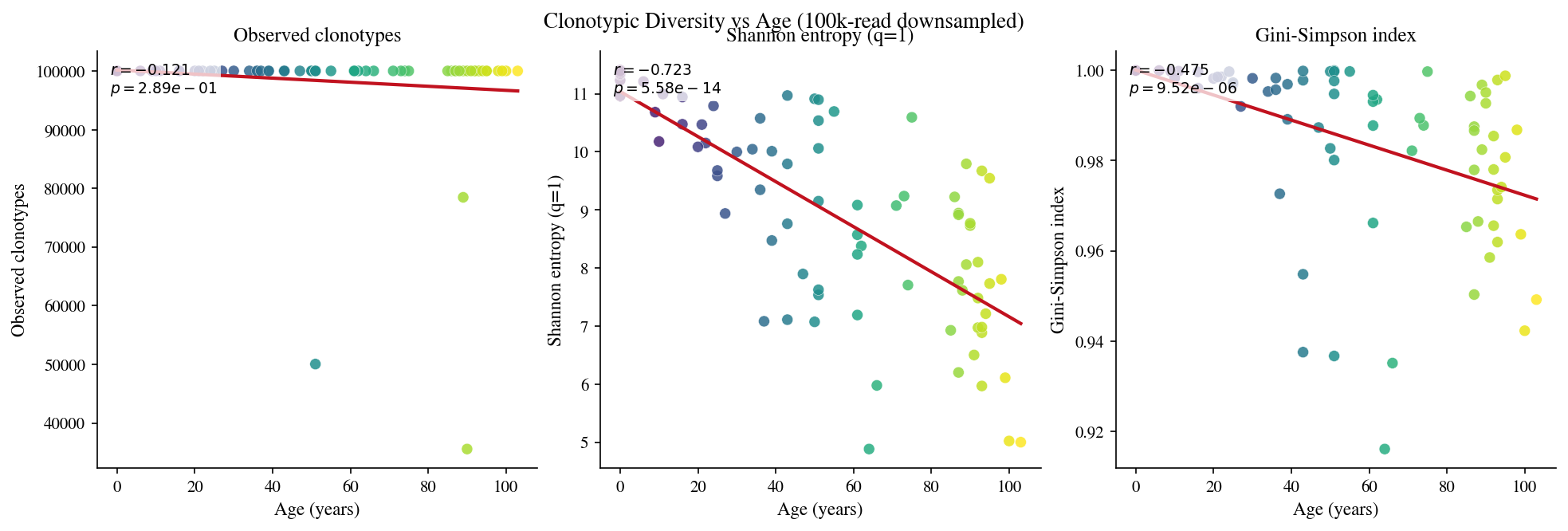
2 · Clonotypic rarefaction#
[7]:
# Cell 7: Clonotypic rarefaction at multiple depths
RARE_DEPTHS = [5_000, 10_000, 25_000, 50_000]
rare_rows = []
for sid, age, rep in zip(sample_ids, ages, reps):
total = int(sum(c.duplicate_count for c in rep.clonotypes))
for m in RARE_DEPTHS:
if m > total:
continue
curve = rarefaction_curve_clonotypes(rep.clonotypes, m_steps=[m], include_exact=True)
row = curve.filter(pl.col('m') == m)
if row.height:
rare_rows.append({'sample_id': sid, 'age': age, 'm': m,
'D': float(row['s_obs'][0])})
rare_df = pl.DataFrame(rare_rows)
fig, axes = plt.subplots(1, len(RARE_DEPTHS), figsize=(14, 4), constrained_layout=True, sharey=False)
for ax, m in zip(axes, RARE_DEPTHS):
sub = rare_df.filter(pl.col('m') == m)
if sub.height == 0:
ax.set_title(f'{m:,} reads\n(no data)')
continue
x = sub['age'].to_numpy().astype(float)
y = sub['D'].to_numpy().astype(float)
ax.scatter(x, y, c=x, cmap='viridis', s=40, alpha=0.85, edgecolors='white', linewidths=0.25)
slope, intercept, r, p, _ = stats.linregress(x, y)
xr = np.linspace(x.min(), x.max(), 200)
ax.plot(xr, slope * xr + intercept, color='#C1121F', lw=2)
ax.set_xlabel('Age (years)')
ax.set_ylabel('Clonotypes')
ax.set_title(f'Rarefaction @ {m:,} reads')
ax.text(0.03, 0.97, f'$r={r:.3f}$', transform=ax.transAxes, va='top', fontsize=9,
bbox=dict(facecolor='white', alpha=0.75, edgecolor='none', pad=2))
fig.suptitle('Clonotypic Rarefaction vs Age', fontsize=13, y=1.02)
plt.show()
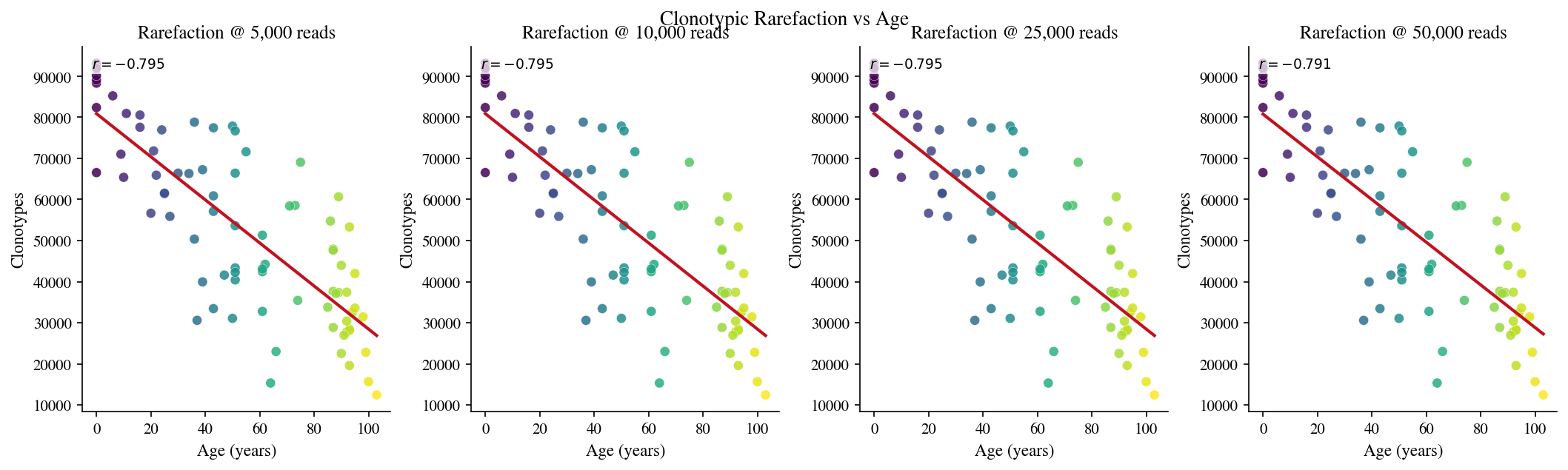
3 · Clonotypic pairwise F overlap#
[8]:
# Cell 8: Clonotypic pairwise F similarity (exact, aavj)
t0 = time.perf_counter()
clono_pairs_pd = pairwise_overlap_matrix(
reps, sample_ids=sample_ids,
metric='exact', threshold=0,
overlap_space='aavj', n_jobs=N_JOBS,
)
EXECUTION_TIMES['clono_pairwise_s'] = time.perf_counter() - t0
clono_pairs = pl.from_pandas(clono_pairs_pd)
print(f'Clonotypic pairwise: {clono_pairs.height:,} pairs in '
f'{EXECUTION_TIMES["clono_pairwise_s"]:.1f}s')
Clonotypic pairwise: 3,081 pairs in 25.8s
[9]:
# Cell 9: Clonotypic donor-vs-pool F overlap
pool_rep = LocusRepertoire(
clonotypes=[c for rep in reps for c in rep.clonotypes],
locus='TRB',
)
print(f'Pool: {pool_rep.clonotype_count:,} clonotypes')
t0 = time.perf_counter()
clono_pool_df = many_vs_pool_overlap(
reps, pool_rep, sample_ids=sample_ids, ages=ages,
metric='exact', threshold=0, overlap_space='aavj', n_jobs=N_JOBS,
)
EXECUTION_TIMES['clono_pool_s'] = time.perf_counter() - t0
print(f'Done in {EXECUTION_TIMES["clono_pool_s"]:.1f}s')
Pool: 4,145,989 clonotypes
Done in 28.2s
4 · Metaclonotype clustering (1mm Hamming + Louvain)#
Each sample is clustered independently. Clonotypes with identical CDR3 amino-acid sequences that differ by at most 1 Hamming substitution are placed in the same cluster (without V/J gene restriction). Singletons (clusters of size 1) are discarded. The Louvain algorithm further merges multi-hop connected dense neighbourhoods.
[10]:
# Cell 10: Per-sample metaclonotype clustering
# ~5–15 s per sample → ~7–20 min total for 79 samples
from mir.common.metaclonotype import MetaClonotypeClustering
meta_clusterings: list[MetaClonotypeClustering] = []
cluster_count_rows = []
t0 = time.perf_counter()
for i, (sid, age, rep) in enumerate(zip(sample_ids, ages, reps)):
t_rep = time.perf_counter()
graph = build_edit_distance_graph(
rep.clonotypes,
metric='hamming',
threshold=METACLONOTYPE_THRESHOLD,
n_jobs=N_JOBS,
)
mc = metaclonotypes_from_graph_communities(
graph,
method=METACLONOTYPE_METHOD,
min_cluster_size=METACLONOTYPE_MIN_SIZE,
)
meta_clusterings.append(mc)
cluster_count_rows.append({
'sample_id': sid, 'age': age,
'n_clonotypes': rep.clonotype_count,
'n_clusters': mc.n_clusters,
'cluster_s': round(time.perf_counter() - t_rep, 2),
})
if (i + 1) % 10 == 0:
elapsed = time.perf_counter() - t0
print(f' [{i+1}/{len(reps)}] {elapsed:.0f}s elapsed — '
f'last cluster: {mc.n_clusters} groups in {cluster_count_rows[-1]["cluster_s"]:.1f}s')
EXECUTION_TIMES['meta_cluster_s'] = time.perf_counter() - t0
cluster_df = pl.DataFrame(cluster_count_rows)
print(f'\nClustered {len(reps)} samples in {EXECUTION_TIMES["meta_cluster_s"]:.1f}s')
print(f'Clusters per sample: '
f'min={cluster_df["n_clusters"].min()} '
f'median={int(cluster_df["n_clusters"].median())} '
f'max={cluster_df["n_clusters"].max()}')
Skipping 1814 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1814 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1814 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1814 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1043 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1043 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1043 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1199 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1199 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1199 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1901 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1901 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1901 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2062 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2062 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2062 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1489 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1489 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1489 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1191 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1191 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1191 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1175 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1175 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1175 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2043 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2043 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2043 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2043 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2161 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2161 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2161 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2161 sequences with non-canonical amino acids (*, _, or non-standard chars)
[10/79] 197s elapsed — last cluster: 2891 groups in 23.4s
Skipping 2071 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2071 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2071 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2071 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1976 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1976 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1976 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1976 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2092 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2092 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2092 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2092 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2049 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2049 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2049 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1132 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1132 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1132 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2278 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2278 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2278 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2278 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1898 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1898 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1898 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1378 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1378 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1378 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1289 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1289 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1289 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 725 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 725 sequences with non-canonical amino acids (*, _, or non-standard chars)
[20/79] 389s elapsed — last cluster: 1427 groups in 7.0s
Skipping 1005 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1005 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 982 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 982 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 982 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1813 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1813 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1813 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1813 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1572 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1572 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1572 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 853 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 853 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 707 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 707 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 499 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 499 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1143 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1143 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1143 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1380 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1380 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1380 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1233 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1233 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1233 sequences with non-canonical amino acids (*, _, or non-standard chars)
[30/79] 502s elapsed — last cluster: 1743 groups in 10.2s
Skipping 896 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 896 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1104 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1104 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1104 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1479 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1479 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1479 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1154 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1154 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1154 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 836 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 836 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1551 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1551 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1551 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1473 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1473 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1473 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2167 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2167 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2167 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2167 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2007 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2007 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2007 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2007 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1542 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1542 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1542 sequences with non-canonical amino acids (*, _, or non-standard chars)
[40/79] 653s elapsed — last cluster: 2396 groups in 16.4s
Skipping 2197 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2197 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2197 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2197 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 922 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 922 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 730 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 730 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 963 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 963 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 963 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 568 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 568 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 945 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 945 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 408 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 408 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 837 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 837 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1215 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1215 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1215 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 826 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 826 sequences with non-canonical amino acids (*, _, or non-standard chars)
[50/79] 743s elapsed — last cluster: 1404 groups in 7.3s
Skipping 1659 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1659 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1659 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 816 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 816 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1991 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1991 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1991 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1991 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1174 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1174 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2553 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2553 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2553 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2553 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 443 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 443 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2490 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2490 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2490 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2490 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 4258 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 4258 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 4258 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 4258 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 3193 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 3193 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 3193 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 3193 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2834 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2834 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2834 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2834 sequences with non-canonical amino acids (*, _, or non-standard chars)
[60/79] 932s elapsed — last cluster: 2907 groups in 30.1s
Skipping 3002 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 3002 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 3002 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 3002 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2752 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2752 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2752 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2752 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 4296 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 4296 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 4296 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 4296 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 4373 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 4373 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 4373 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 4373 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 3766 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 3766 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 3766 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 3766 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1690 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1690 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1690 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 988 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 988 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1142 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1142 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1142 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 830 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 830 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 786 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 786 sequences with non-canonical amino acids (*, _, or non-standard chars)
[70/79] 1142s elapsed — last cluster: 1671 groups in 8.7s
Skipping 1668 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1668 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1668 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1512 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1512 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1512 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 949 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 949 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 949 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1087 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1087 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1087 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 774 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 774 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 947 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 947 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1014 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1014 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1014 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1359 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1359 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1359 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1218 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1218 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1218 sequences with non-canonical amino acids (*, _, or non-standard chars)
Clustered 79 samples in 1257.1s
Clusters per sample: min=633 median=2189 max=3271
[11]:
# Cell 11: Cluster count and compression ratio vs age
cluster_df = cluster_df.with_columns(
(pl.col('n_clusters') / pl.col('n_clonotypes')).alias('compression')
)
fig, axes = plt.subplots(1, 2, figsize=(11, 4.2), constrained_layout=True)
for ax, (col, label) in zip(axes, [
('n_clusters', 'Number of metaclonotypes'),
('compression', 'Metaclonotypes / clonotypes'),
]):
x = cluster_df['age'].to_numpy().astype(float)
y = cluster_df[col].to_numpy().astype(float)
ax.scatter(x, y, c=x, cmap='viridis', s=48, alpha=0.85, edgecolors='white', linewidths=0.25)
slope, intercept, r, p, _ = stats.linregress(x, y)
xr = np.linspace(x.min(), x.max(), 200)
ax.plot(xr, slope * xr + intercept, color='#C1121F', lw=2)
ax.set_xlabel('Age (years)')
ax.set_ylabel(label)
ax.set_title(label)
ax.text(0.03, 0.97, f'$r={r:.3f}$\n$p={p:.2e}$',
transform=ax.transAxes, va='top', fontsize=9,
bbox=dict(facecolor='white', alpha=0.75, edgecolor='none', pad=2))
fig.suptitle('Metaclonotype Cluster Count vs Age', fontsize=13, y=1.02)
plt.show()
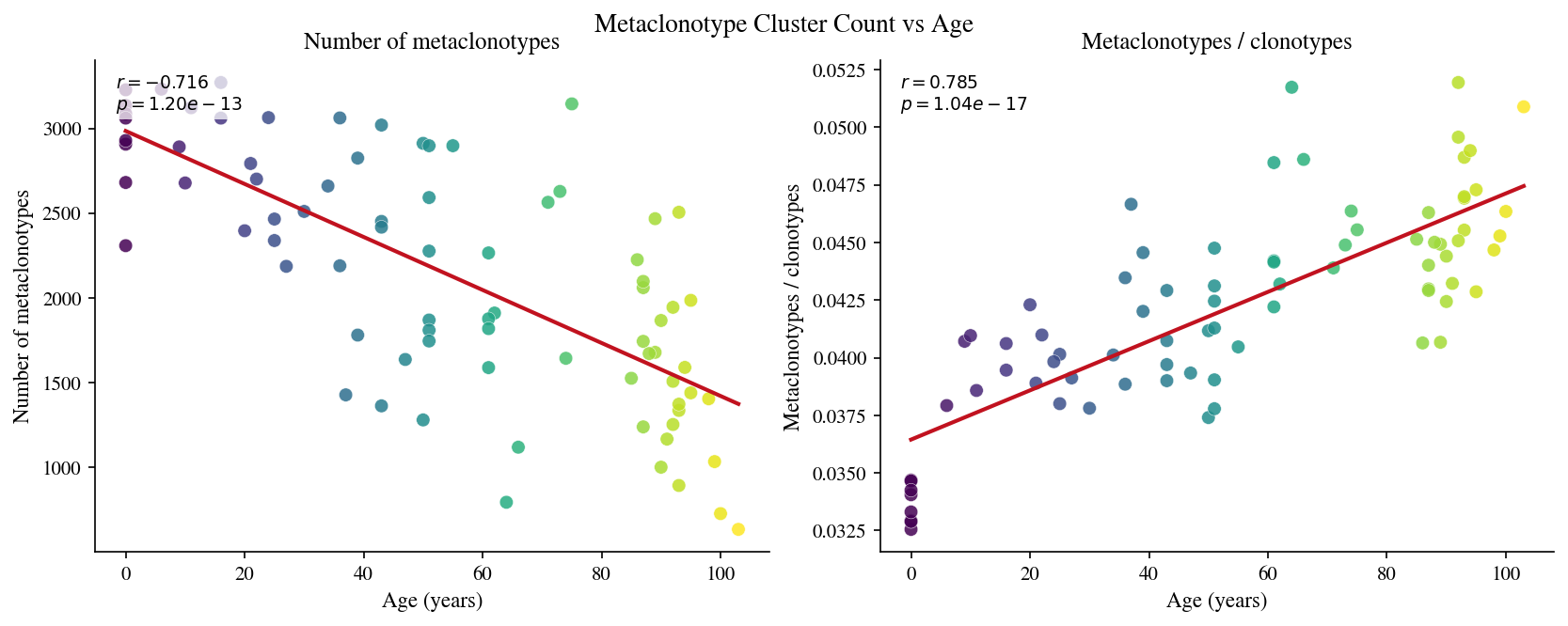
5 · Functional diversity#
[12]:
# Cell 12: Functional diversity per sample (Shannon at metaclonotype level)
func_div_rows = []
for sid, age, rep, mc in zip(sample_ids, ages, reps, meta_clusterings):
if mc.n_clusters == 0:
continue
d = functional_diversity(rep, mc)
func_div_rows.append({
'sample_id': sid, 'age': age,
'func_shannon': d.shannon,
'func_gini_simpson': d.gini_simpson,
'func_chao1': d.chao1,
})
func_div_df = pl.DataFrame(func_div_rows)
fig, axes = plt.subplots(1, 3, figsize=(13, 4.2), constrained_layout=True)
for ax, (col, label) in zip(axes, [
('func_shannon', 'Functional Shannon (q=1)'),
('func_gini_simpson', 'Functional Gini-Simpson'),
('func_chao1', 'Functional Chao1'),
]):
x = func_div_df['age'].to_numpy().astype(float)
y = func_div_df[col].to_numpy().astype(float)
ax.scatter(x, y, c=x, cmap='viridis', s=45, alpha=0.85, edgecolors='white', linewidths=0.25)
slope, intercept, r, p, _ = stats.linregress(x, y)
xr = np.linspace(x.min(), x.max(), 200)
ax.plot(xr, slope * xr + intercept, color='#C1121F', lw=2)
ax.set_xlabel('Age (years)')
ax.set_ylabel(label)
ax.set_title(label)
ax.text(0.03, 0.97, f'$r={r:.3f}$\n$p={p:.2e}$',
transform=ax.transAxes, va='top', fontsize=9,
bbox=dict(facecolor='white', alpha=0.75, edgecolor='none', pad=2))
fig.suptitle('Functional Diversity vs Age (1mm Hamming + Louvain metaclonotypes)', fontsize=13, y=1.02)
plt.show()
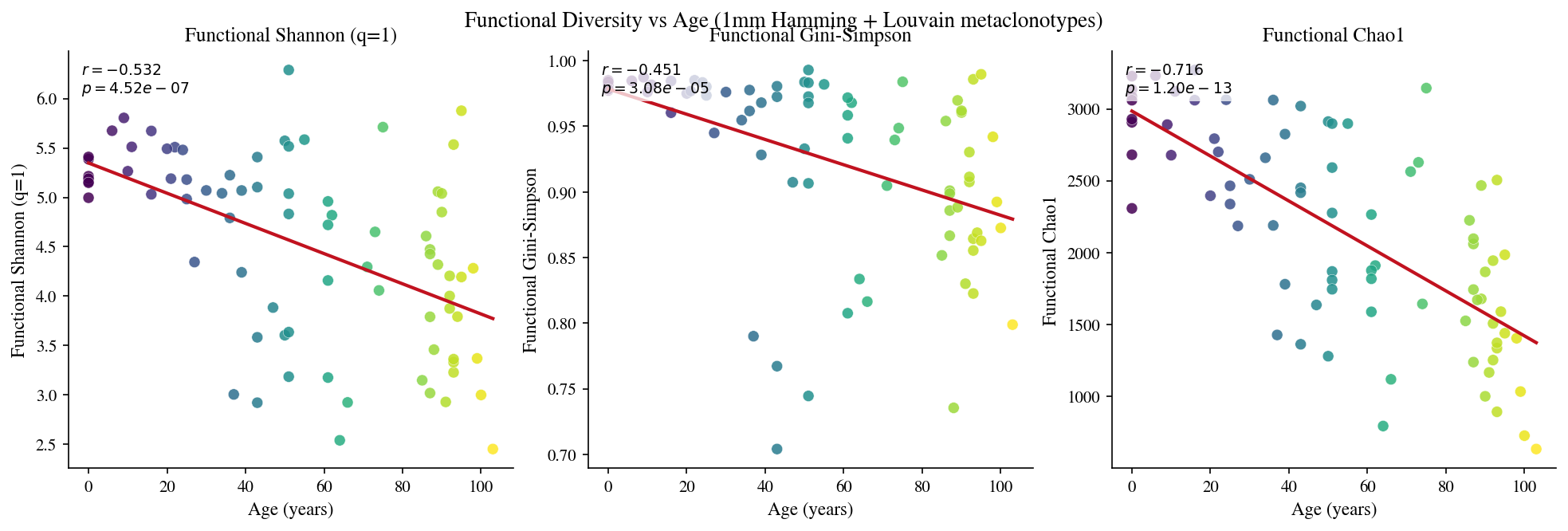
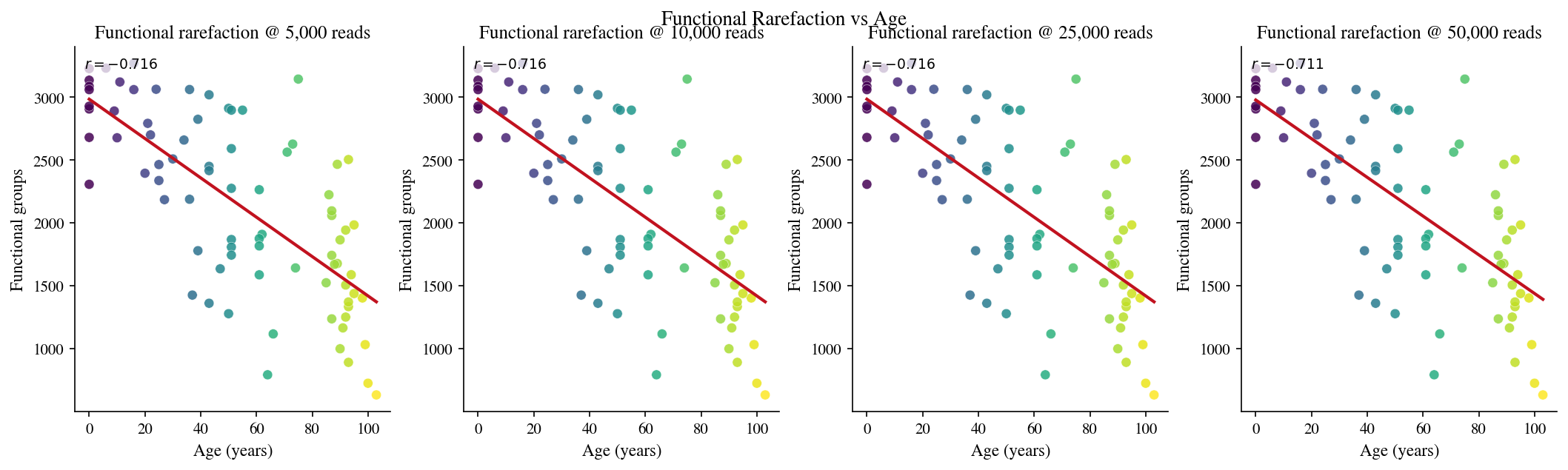
6 · Functional rarefaction#
[13]:
# Cell 13: Functional rarefaction at multiple depths
FUNC_RARE_DEPTHS = [5_000, 10_000, 25_000, 50_000]
func_rare_rows = []
for sid, age, rep, mc in zip(sample_ids, ages, reps, meta_clusterings):
if mc.n_clusters == 0:
continue
total = int(sum(c.duplicate_count for c in rep.clonotypes))
valid_depths = [m for m in FUNC_RARE_DEPTHS if m <= total]
if not valid_depths:
continue
try:
curve = functional_rarefaction_curve(rep, mc, m_steps=valid_depths, include_exact=False)
for m in valid_depths:
row = curve.filter(pl.col('m') == m)
if row.height:
func_rare_rows.append({
'sample_id': sid, 'age': age, 'm': m,
'D': float(row['s_obs'][0]),
})
except Exception:
pass
func_rare_df = pl.DataFrame(func_rare_rows)
fig, axes = plt.subplots(1, len(FUNC_RARE_DEPTHS), figsize=(14, 4), constrained_layout=True)
for ax, m in zip(axes, FUNC_RARE_DEPTHS):
sub = func_rare_df.filter(pl.col('m') == m)
if sub.height < 2:
ax.set_title(f'{m:,} reads\n(insufficient data)')
continue
x = sub['age'].to_numpy().astype(float)
y = sub['D'].to_numpy().astype(float)
ax.scatter(x, y, c=x, cmap='viridis', s=40, alpha=0.85, edgecolors='white', linewidths=0.25)
slope, intercept, r, p, _ = stats.linregress(x, y)
xr = np.linspace(x.min(), x.max(), 200)
ax.plot(xr, slope * xr + intercept, color='#C1121F', lw=2)
ax.set_xlabel('Age (years)')
ax.set_ylabel('Functional groups')
ax.set_title(f'Functional rarefaction @ {m:,} reads')
ax.text(0.03, 0.97, f'$r={r:.3f}$', transform=ax.transAxes, va='top', fontsize=9,
bbox=dict(facecolor='white', alpha=0.75, edgecolor='none', pad=2))
fig.suptitle('Functional Rarefaction vs Age', fontsize=13, y=1.02)
plt.show()
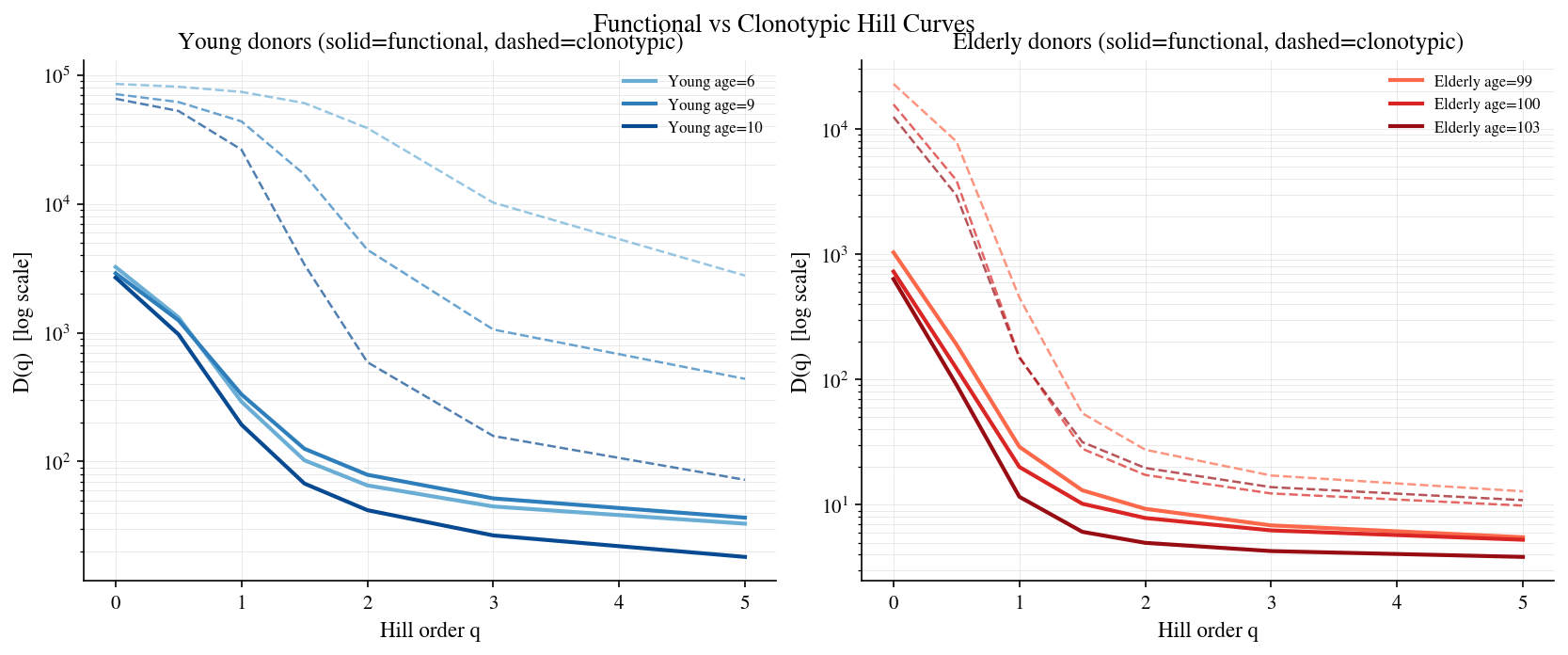
[14]:
# Cell 14: Functional Hill curves for young vs elderly donors
# Pick 3 youngest and 3 oldest samples (exclude cord-blood age=0)
age_sorted = sorted(
[(age, i) for i, age in enumerate(ages) if age > 0],
key=lambda t: t[0]
)
young_idx = [i for _, i in age_sorted[:3]]
old_idx = [i for _, i in age_sorted[-3:]]
q_values = [0.0, 0.5, 1.0, 1.5, 2.0, 3.0, 5.0]
fig, axes = plt.subplots(1, 2, figsize=(11, 4.4), constrained_layout=True)
colors_young = plt.cm.Blues(np.linspace(0.5, 0.9, 3))
colors_old = plt.cm.Reds(np.linspace(0.5, 0.9, 3))
for ax, (group_idx, colors, group_label) in zip(
axes,
[(young_idx, colors_young, 'Young'), (old_idx, colors_old, 'Elderly')]
):
for j, (idx, col) in enumerate(zip(group_idx, colors)):
rep = reps[idx]
mc = meta_clusterings[idx]
age = ages[idx]
if mc.n_clusters < 2:
continue
# Clonotypic Hill (dashed) — column is 'hill'
hill_c = hill_curve_clonotypes(rep.clonotypes, q_values=q_values)
ax.plot(hill_c['q'], hill_c['hill'], '--', color=col, lw=1.2, alpha=0.7)
# Functional Hill (solid) — column is 'hill'
hill_f = functional_hill_curve(rep, mc, q_values=q_values)
ax.plot(hill_f['q'], hill_f['hill'], '-', color=col, lw=2,
label=f'{group_label} age={age}')
ax.set_yscale('log')
ax.set_xlabel('Hill order q')
ax.set_ylabel('D(q) [log scale]')
ax.set_title(f'{group_label} donors (solid=functional, dashed=clonotypic)')
ax.legend(fontsize=8, frameon=False)
ax.grid(alpha=0.3, which='both')
fig.suptitle('Functional vs Clonotypic Hill Curves', fontsize=13, y=1.02)
plt.show()
7 · Functional pairwise F overlap#
The functional F overlap between two samples A and B is defined as:
where \(|\text{shared}_A|\) is the number of metaclonotype groups in A that share at least one clonotype identity with any group in B (and vice versa). This is the F1-score (harmonic mean) of the two asymmetric sharing fractions.
[15]:
# Cell 15: Functional pairwise F overlap
# O(N^2) calls to functional_overlap_1 — fast because identity sets are cached per sample.
n = len(reps)
func_pair_rows = []
t0 = time.perf_counter()
for i in range(n):
rep_a, mc_a, sid_a, age_a = reps[i], meta_clusterings[i], sample_ids[i], ages[i]
if mc_a.n_clusters == 0:
continue
for j in range(i + 1, n):
rep_b, mc_b, sid_b, age_b = reps[j], meta_clusterings[j], sample_ids[j], ages[j]
if mc_b.n_clusters == 0:
continue
r = functional_overlap_1(rep_a, mc_a, rep_b, mc_b)
sa = float(r['a_shared_clusters'][0])
sb = float(r['b_shared_clusters'][0])
ca = float(r['a_clusters'][0])
cb = float(r['b_clusters'][0])
denom = ca * sb + cb * sa
f_func = 2 * sa * sb / denom if denom > 0 else 0.0
func_pair_rows.append({
'sample_id_1': sid_a, 'sample_id_2': sid_b,
'age1': age_a, 'age2': age_b,
'age_diff': abs(age_a - age_b),
'f_func': f_func,
'frac_a': sa / ca if ca > 0 else 0.0,
'frac_b': sb / cb if cb > 0 else 0.0,
})
EXECUTION_TIMES['func_pairwise_s'] = time.perf_counter() - t0
func_pair_df = pl.DataFrame(func_pair_rows)
print(f'Functional pairwise: {func_pair_df.height:,} pairs in '
f'{EXECUTION_TIMES["func_pairwise_s"]:.1f}s')
Functional pairwise: 3,081 pairs in 167.9s
[16]:
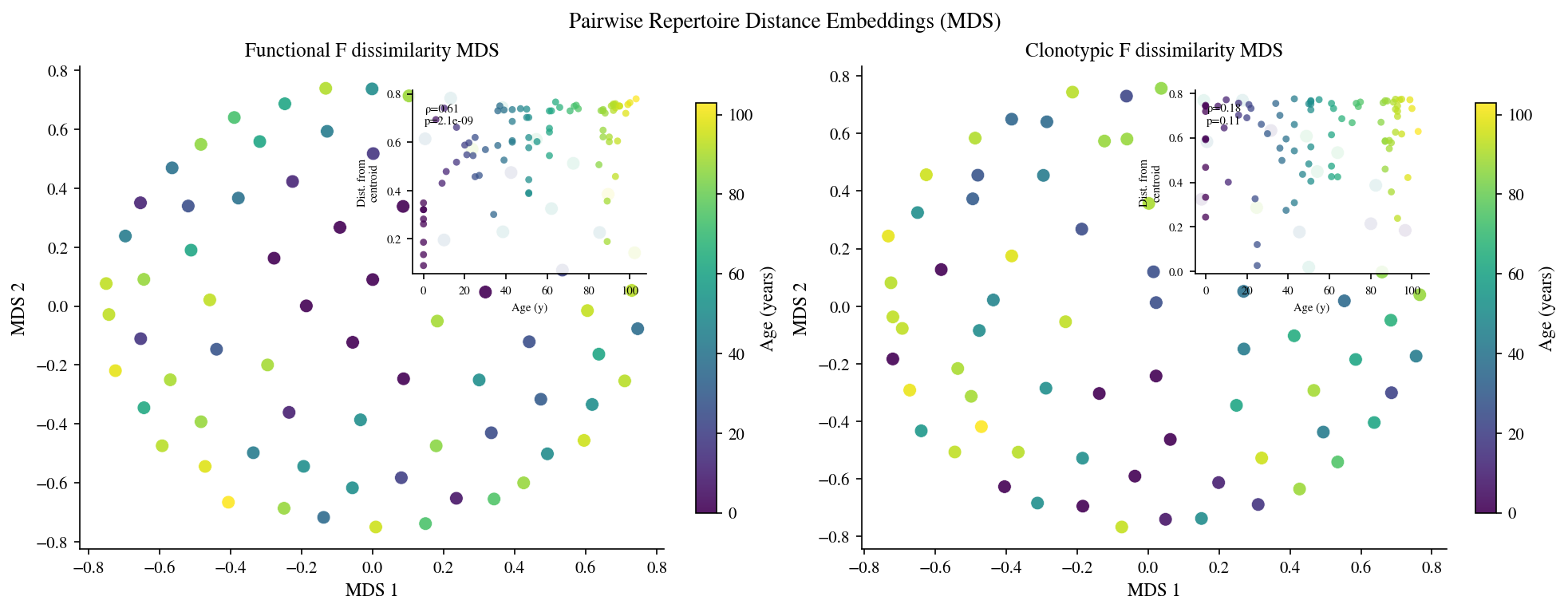
# Cell 16: MDS embedding of pairwise functional and clonotypic dissimilarities
# Distances are defined as dissimilarity = 1 − F_similarity (functional) or
# 1 − f_similarity (clonotypic). MDS preserves pairwise distance structure.
from sklearn.manifold import MDS
def _pairs_to_dist_matrix(pairs_df, f_col: str, ids: list) -> np.ndarray:
n = len(ids)
idx = {sid: i for i, sid in enumerate(ids)}
mat = np.zeros((n, n))
for row in pairs_df.iter_rows(named=True):
i = idx.get(row['sample_id_1'])
j = idx.get(row['sample_id_2'])
if i is None or j is None:
continue
v = row[f_col]
d = 1.0 - (float(v) if v is not None else 0.0)
mat[i, j] = d
mat[j, i] = d
return mat
func_dist = _pairs_to_dist_matrix(func_pair_df, 'f_func', sample_ids)
clono_dist = _pairs_to_dist_matrix(clono_pairs, 'f_similarity', sample_ids)
_mds_kw = dict(
n_components=2, dissimilarity='precomputed',
normalized_stress='auto', random_state=42, n_init=4, max_iter=400,
)
mds_func = MDS(**_mds_kw).fit_transform(func_dist)
mds_clono = MDS(**_mds_kw).fit_transform(clono_dist)
age_arr = np.array(ages, dtype=float)
fig, axes = plt.subplots(1, 2, figsize=(13, 5), constrained_layout=True)
for ax, emb, title in zip(
axes,
[mds_func, mds_clono],
['Functional F dissimilarity MDS', 'Clonotypic F dissimilarity MDS'],
):
sc = ax.scatter(
emb[:, 0], emb[:, 1],
c=age_arr, cmap='viridis', s=64, alpha=0.9,
linewidths=0.3, edgecolors='white',
)
plt.colorbar(sc, ax=ax, label='Age (years)', shrink=0.85)
ax.set_xlabel('MDS 1', fontsize=11)
ax.set_ylabel('MDS 2', fontsize=11)
ax.set_title(title, fontsize=12)
# Inset: distance from MDS centroid vs age (Spearman ρ)
centroid = emb.mean(axis=0)
dist_from_c = np.linalg.norm(emb - centroid, axis=1)
rho, pval = stats.spearmanr(age_arr, dist_from_c)
axins = ax.inset_axes([0.57, 0.57, 0.40, 0.38])
axins.scatter(age_arr, dist_from_c, c=age_arr, cmap='viridis',
s=18, alpha=0.75, edgecolors='none')
axins.set_xlabel('Age (y)', fontsize=7)
axins.set_ylabel('Dist. from\ncentroid', fontsize=7)
axins.tick_params(labelsize=7)
axins.text(0.05, 0.93, f'ρ={rho:.2f}\np={pval:.2g}',
transform=axins.transAxes, fontsize=7, va='top')
axins.set_facecolor('white')
axins.patch.set_alpha(0.88)
fig.suptitle('Pairwise Repertoire Distance Embeddings (MDS)', fontsize=13)
plt.show()
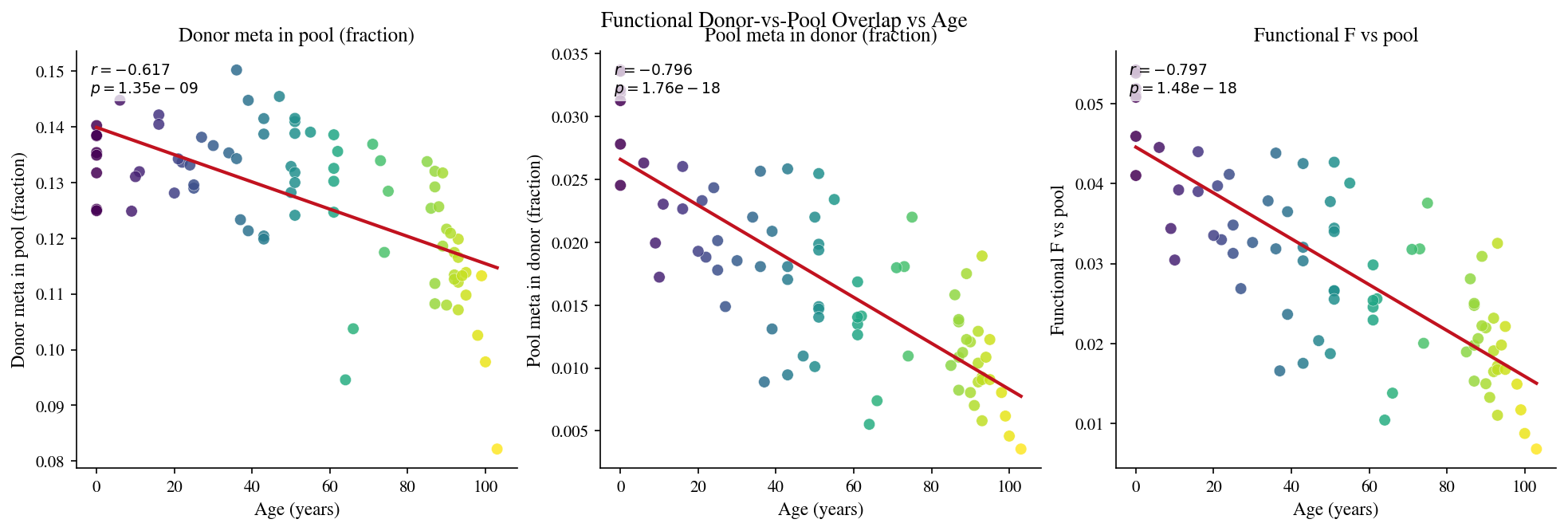
8 · Functional donor-vs-pool F overlap#
Build metaclonotype clusters for the pooled repertoire; then for each donor compute the fraction of donor metaclonotypes that share identity with a pool metaclonotype.
[17]:
# Cell 17: Cluster the pooled repertoire
# The raw pool has duplicates (same CDR3+V+J from multiple donors).
# Deduplicate by (junction_aa, v_gene, j_gene) before graph building.
from mir.common.sampling import downsample as _downsample
_key = lambda c: (c.junction_aa, c.v_gene or '', c.j_gene or '')
_seen: dict = {}
for c in pool_rep.clonotypes:
k = _key(c)
if k not in _seen:
_seen[k] = c
pool_dedup_clonotypes = list(_seen.values())
MAX_POOL_GRAPH = 300_000
if len(pool_dedup_clonotypes) > MAX_POOL_GRAPH:
import random as _rng
_rng.seed(42)
pool_dedup_clonotypes = _rng.sample(pool_dedup_clonotypes, MAX_POOL_GRAPH)
print(f'Pool: {pool_rep.clonotype_count:,} raw → {MAX_POOL_GRAPH:,} unique (sampled)')
else:
print(f'Pool: {pool_rep.clonotype_count:,} raw → {len(pool_dedup_clonotypes):,} unique clonotypes')
pool_rep_dedup = LocusRepertoire(clonotypes=pool_dedup_clonotypes, locus='TRB')
t0 = time.perf_counter()
pool_graph = build_edit_distance_graph(
pool_rep_dedup.clonotypes,
metric='hamming',
threshold=METACLONOTYPE_THRESHOLD,
n_jobs=N_JOBS,
)
meta_pool = metaclonotypes_from_graph_communities(
pool_graph,
method=METACLONOTYPE_METHOD,
min_cluster_size=METACLONOTYPE_MIN_SIZE,
)
EXECUTION_TIMES['pool_cluster_s'] = time.perf_counter() - t0
print(f'Pool metaclonotypes: {meta_pool.n_clusters:,} in {EXECUTION_TIMES["pool_cluster_s"]:.1f}s')
Pool: 4,145,989 raw → 300,000 unique (sampled)
Skipping 9717 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 9717 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 9717 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 9717 sequences with non-canonical amino acids (*, _, or non-standard chars)
Pool metaclonotypes: 10,679 in 312.9s
[18]:
# Cell 18: Functional donor-vs-pool overlap
func_pool_rows = []
t0 = time.perf_counter()
for sid, age, rep, mc in zip(sample_ids, ages, reps, meta_clusterings):
if mc.n_clusters == 0:
continue
r = functional_overlap_1(rep, mc, pool_rep_dedup, meta_pool)
frac_donor = float(r['a_overlap_fraction'][0]) # fraction of donor meta shared with pool
frac_pool = float(r['b_overlap_fraction'][0]) # fraction of pool meta shared with donor
denom = frac_donor + frac_pool
f_func = 2 * frac_donor * frac_pool / denom if denom > 0 else 0.0
func_pool_rows.append({
'sample_id': sid, 'age': age,
'frac_donor_in_pool': frac_donor,
'frac_pool_in_donor': frac_pool,
'f_func_pool': f_func,
})
EXECUTION_TIMES['func_pool_s'] = time.perf_counter() - t0
func_pool_df = pl.DataFrame(func_pool_rows)
print(f'Functional vs-pool: {len(func_pool_rows)} samples in {EXECUTION_TIMES["func_pool_s"]:.1f}s')
fig, axes = plt.subplots(1, 3, figsize=(13, 4.2), constrained_layout=True)
for ax, (col, label) in zip(axes, [
('frac_donor_in_pool', 'Donor meta in pool (fraction)'),
('frac_pool_in_donor', 'Pool meta in donor (fraction)'),
('f_func_pool', 'Functional F vs pool'),
]):
x = func_pool_df['age'].to_numpy().astype(float)
y = func_pool_df[col].to_numpy().astype(float)
ax.scatter(x, y, c=x, cmap='viridis', s=48, alpha=0.85, edgecolors='white', linewidths=0.25)
slope, intercept, r, p, _ = stats.linregress(x, y)
xr = np.linspace(x.min(), x.max(), 200)
ax.plot(xr, slope * xr + intercept, color='#C1121F', lw=2)
ax.set_xlabel('Age (years)')
ax.set_ylabel(label)
ax.set_title(label)
ax.text(0.03, 0.97, f'$r={r:.3f}$\n$p={p:.2e}$',
transform=ax.transAxes, va='top', fontsize=9,
bbox=dict(facecolor='white', alpha=0.75, edgecolor='none', pad=2))
fig.suptitle('Functional Donor-vs-Pool Overlap vs Age', fontsize=13, y=1.02)
plt.show()
Functional vs-pool: 79 samples in 22.9s
9 · Side-by-side: clonotypic vs functional metrics#
[19]:
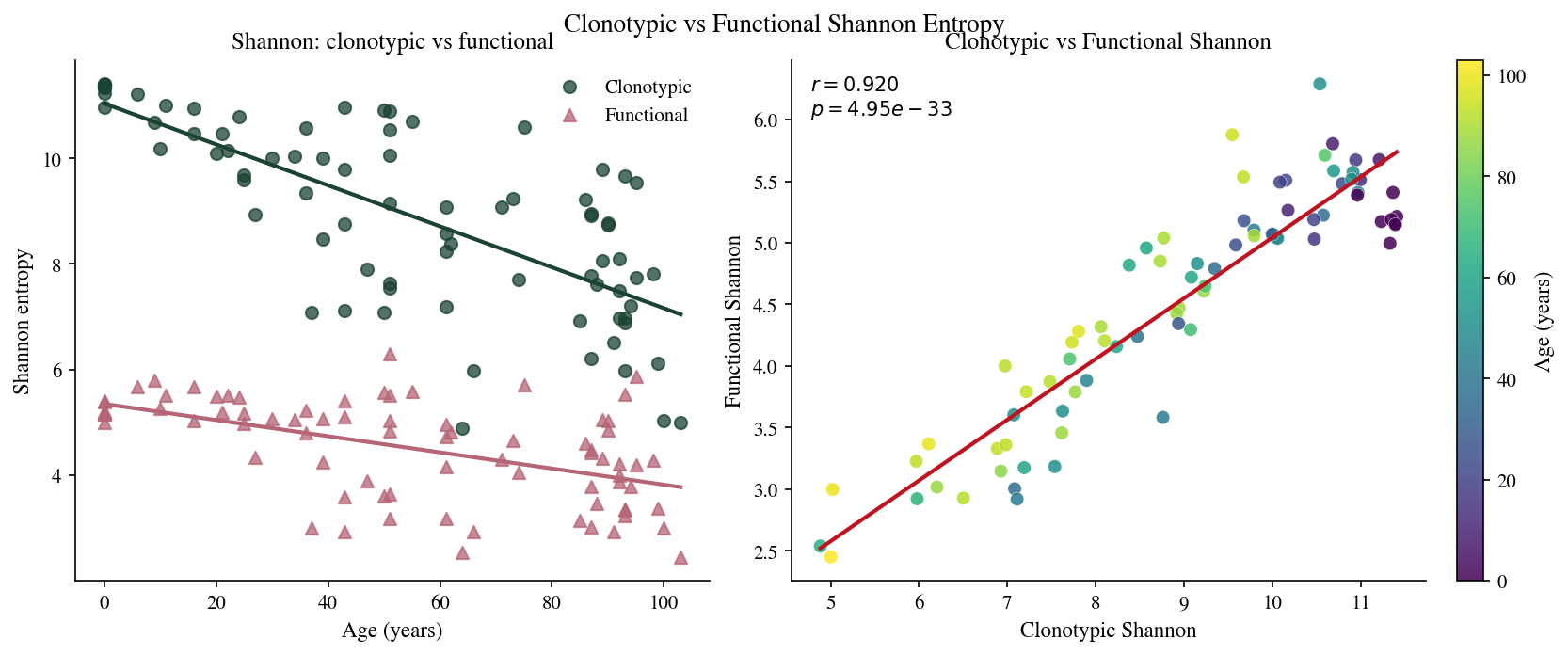
# Cell 19: Clonotypic vs functional Shannon entropy vs age
merged = clono_div_df.join(
func_div_df.select(['sample_id', 'func_shannon']),
on='sample_id', how='inner',
)
fig, axes = plt.subplots(1, 2, figsize=(11, 4.4), constrained_layout=True)
ax = axes[0]
x = merged['age'].to_numpy().astype(float)
yc = merged['shannon'].to_numpy().astype(float)
yf = merged['func_shannon'].to_numpy().astype(float)
ax.scatter(x, yc, s=40, alpha=0.75, color='#1B4332', label='Clonotypic')
ax.scatter(x, yf, s=40, alpha=0.75, color='#B56576', marker='^', label='Functional')
for col, color in [('shannon', '#1B4332'), ('func_shannon', '#B56576')]:
y = merged[col].to_numpy().astype(float)
slope, intercept, _, _, _ = stats.linregress(x, y)
ax.plot(np.linspace(x.min(), x.max(), 200),
slope * np.linspace(x.min(), x.max(), 200) + intercept,
color=color, lw=2)
ax.set_xlabel('Age (years)')
ax.set_ylabel('Shannon entropy')
ax.set_title('Shannon: clonotypic vs functional')
ax.legend(frameon=False)
# Correlation between clonotypic and functional Shannon
ax2 = axes[1]
ax2.scatter(yc, yf, c=x, cmap='viridis', s=48, alpha=0.85, edgecolors='white', linewidths=0.25)
slope2, intercept2, r2, p2, _ = stats.linregress(yc, yf)
xr2 = np.linspace(yc.min(), yc.max(), 200)
ax2.plot(xr2, slope2 * xr2 + intercept2, color='#C1121F', lw=2)
ax2.set_xlabel('Clonotypic Shannon')
ax2.set_ylabel('Functional Shannon')
ax2.set_title('Clonotypic vs Functional Shannon')
ax2.text(0.03, 0.97, f'$r={r2:.3f}$\n$p={p2:.2e}$',
transform=ax2.transAxes, va='top', fontsize=10,
bbox=dict(facecolor='white', alpha=0.75, edgecolor='none', pad=2))
plt.colorbar(ax2.collections[0], ax=ax2, label='Age (years)')
fig.suptitle('Clonotypic vs Functional Shannon Entropy', fontsize=13, y=1.02)
plt.show()
[20]:
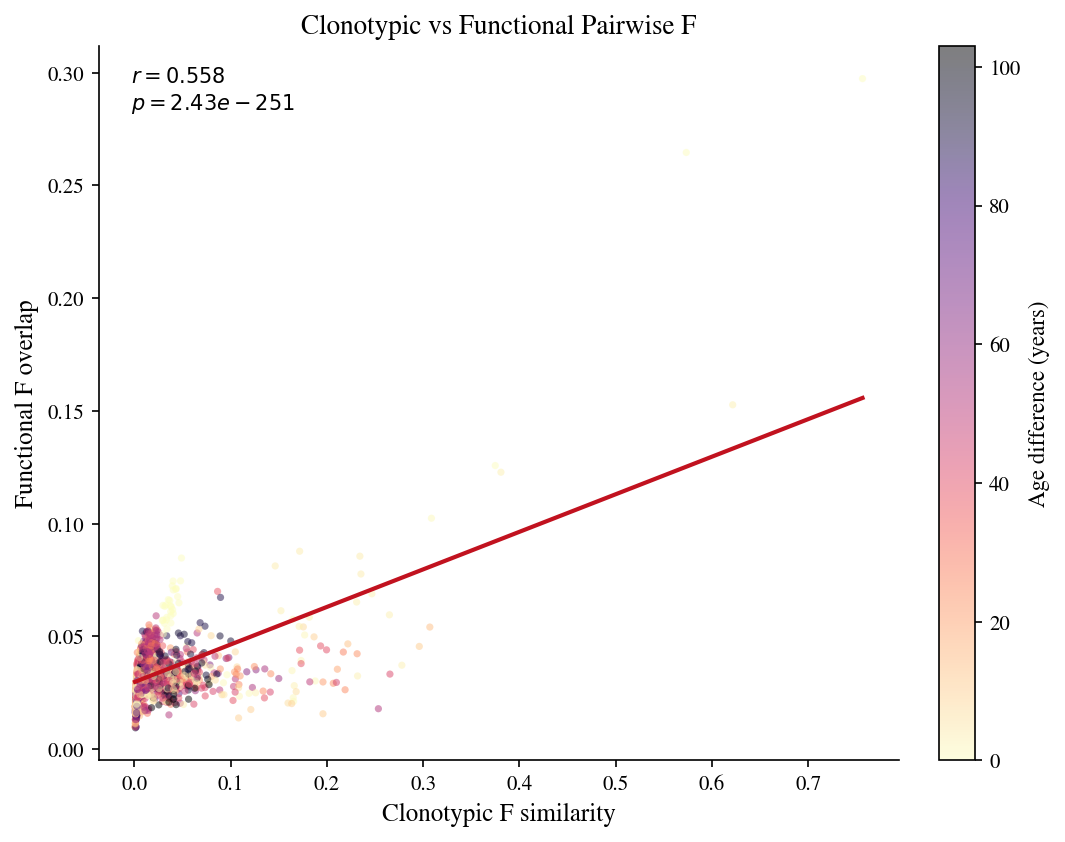
# Cell 20: Clonotypic vs functional pairwise F — correlation scatter
# 'combined' retains age_diff for the Spearman summary cell that follows.
clono_f = (
clono_pairs
.with_columns(pl.col('f_similarity').alias('f_clono'))
.select(['sample_id_1', 'sample_id_2', 'f_clono'])
)
combined = clono_f.join(
func_pair_df.select(['sample_id_1', 'sample_id_2', 'age_diff', 'f_func']),
on=['sample_id_1', 'sample_id_2'],
how='inner',
)
fc = combined['f_clono'].to_numpy().astype(float)
ff = combined['f_func'].to_numpy().astype(float)
age_diff = combined['age_diff'].to_numpy().astype(float)
fig, ax = plt.subplots(figsize=(7, 5.5), constrained_layout=True)
sc = ax.scatter(fc, ff, c=age_diff, cmap='magma_r', s=12, alpha=0.5, edgecolors='none')
slope, intercept, r, p, _ = stats.linregress(fc, ff)
xr = np.linspace(fc.min(), fc.max(), 200)
ax.plot(xr, slope * xr + intercept, color='#C1121F', lw=2)
ax.set_xlabel('Clonotypic F similarity', fontsize=12)
ax.set_ylabel('Functional F overlap', fontsize=12)
ax.set_title('Clonotypic vs Functional Pairwise F', fontsize=13)
ax.text(0.04, 0.97, f'$r={r:.3f}$\n$p={p:.2e}$',
transform=ax.transAxes, va='top', fontsize=10,
bbox=dict(facecolor='white', alpha=0.75, edgecolor='none', pad=2))
plt.colorbar(sc, ax=ax, label='Age difference (years)')
plt.show()
10 · Age correlation summary#
[21]:
# Cell 21: Spearman correlations with age — all metrics
import polars as pl
summary_rows = []
# Per-sample metrics
combined_per_sample = (
clono_div_df
.join(func_div_df.select(['sample_id', 'func_shannon', 'func_gini_simpson', 'func_chao1']),
on='sample_id', how='inner')
.join(cluster_df.select(['sample_id', 'n_clusters', 'compression']),
on='sample_id', how='left')
.join(func_pool_df.select(['sample_id', 'frac_donor_in_pool', 'f_func_pool']),
on='sample_id', how='left')
)
age_vec = combined_per_sample['age'].to_numpy().astype(float)
for col, label in [
('n_clonotypes', 'Clonotypic richness (observed)'),
('shannon', 'Clonotypic Shannon'),
('gini_simpson', 'Clonotypic Gini-Simpson'),
('chao1', 'Clonotypic Chao1'),
('n_clusters', 'Metaclonotype count'),
('compression', 'Compression (meta/clono)'),
('func_shannon', 'Functional Shannon'),
('func_gini_simpson', 'Functional Gini-Simpson'),
('func_chao1', 'Functional Chao1'),
('frac_donor_in_pool', 'Func fraction in pool'),
('f_func_pool', 'Functional F vs pool'),
]:
if col not in combined_per_sample.columns:
continue
vals = combined_per_sample[col].to_numpy().astype(float)
mask = np.isfinite(age_vec) & np.isfinite(vals)
if mask.sum() < 3:
continue
rho, pval = stats.spearmanr(age_vec[mask], vals[mask])
summary_rows.append({'metric': label, 'level': 'sample', 'rho': round(rho, 3), 'p_value': round(pval, 4)})
# Pairwise metrics vs age difference
for df, col, label in [
(combined, 'f_clono', 'Clonotypic pairwise F'),
(combined, 'f_func', 'Functional pairwise F'),
]:
if col not in df.columns:
continue
x = df['age_diff'].to_numpy().astype(float)
y = df[col].to_numpy().astype(float)
mask = np.isfinite(x) & np.isfinite(y)
if mask.sum() < 3:
continue
rho, pval = stats.spearmanr(x[mask], y[mask])
summary_rows.append({'metric': label + ' ~ age_diff', 'level': 'pair', 'rho': round(rho, 3), 'p_value': round(pval, 4)})
summary_table = pl.DataFrame(summary_rows).sort('rho')
print('Spearman ρ with age (sample metrics) or age_diff (pairwise metrics):')
print(summary_table.to_pandas().to_string(index=False))
print('\nExecution timing:')
for k, v in EXECUTION_TIMES.items():
print(f' {k:<30} {v:.1f}s')
Spearman ρ with age (sample metrics) or age_diff (pairwise metrics):
metric level rho p_value
Functional F vs pool sample -0.790 0.0000
Clonotypic Shannon sample -0.746 0.0000
Metaclonotype count sample -0.720 0.0000
Functional Chao1 sample -0.720 0.0000
Clonotypic Gini-Simpson sample -0.685 0.0000
Clonotypic Chao1 sample -0.681 0.0000
Func fraction in pool sample -0.640 0.0000
Functional Gini-Simpson sample -0.555 0.0000
Functional Shannon sample -0.537 0.0000
Clonotypic richness (observed) sample -0.127 0.2663
Functional pairwise F ~ age_diff pair -0.081 0.0000
Clonotypic pairwise F ~ age_diff pair -0.009 0.6310
Compression (meta/clono) sample 0.795 0.0000
Execution timing:
asset_refresh_s 0.6s
load_s 108.4s
clono_pairwise_s 25.8s
clono_pool_s 28.2s
meta_cluster_s 1257.1s
func_pairwise_s 167.9s
pool_cluster_s 312.9s
func_pool_s 22.9s