Metaclonotype API Examples#
This notebook demonstrates how to build and analyze metaclonotypes using mirpy.
Metaclonotypes group clonotypes that are functionally similar (share convergent CDR3 sequences or belong to the same antigen-binding family). They allow functional diversity metrics that are more robust to clonal expansion than raw clonotype counts.
Workflow#
Load a real TRB repertoire from the AIRR benchmark.
Build metaclonotypes with the high-level
cluster_metaclonotypesAPI.Summarize and visualize clusters.
Compare functional vs clonotypic diversity metrics.
Build metaclonotypes from ALICE/TCRNET enrichment results.
[1]:
# Environment versions
import platform
import sys
import polars as pl
import importlib.metadata as _meta
print(f'Python {platform.python_version()}')
for pkg in ['mirpy-lib', 'polars', 'numpy', 'igraph']:
try:
print(f' {pkg}: {_meta.version(pkg)}')
except _meta.PackageNotFoundError:
pass
Python 3.12.12
mirpy-lib: 1.1.0
polars: 1.40.1
numpy: 1.26.4
igraph: 1.0.0
[2]:
# Imports and data loading
import time
from pathlib import Path
import matplotlib.pyplot as plt
import numpy as np
import polars as pl
from mir.biomarkers.metaclonotype_cluster import (
MetaclonotypeClusterConfig,
cluster_metaclonotypes,
)
from mir.biomarkers.alice import add_alice_metadata
from mir.common.diversity import summarize_clonotypes, hill_curve_clonotypes
from mir.common.metaclonotype import (
functional_diversity,
functional_hill_curve,
summarize_metaclonotypes,
)
from mir.common.parser import VDJtoolsParser
from mir.common.repertoire import LocusRepertoire
from mir.common.sampling import downsample
from mir.utils.notebook_assets import ensure_airr_benchmark
plt.rcParams.update({
'figure.dpi': 150,
'font.size': 10,
'axes.spines.top': False,
'axes.spines.right': False,
})
# Download AIRR benchmark data on first run (cached after that)
benchmark_root = ensure_airr_benchmark(allow_patterns=['vdjtools/**'])
vdjtools_dir = benchmark_root / 'vdjtools'
# Load one aging-cohort TRB sample and downsample to 20k reads for speed
sample_path = vdjtools_dir / 'A3-i101.txt.gz'
clonotypes = VDJtoolsParser(sep='\t').parse(str(sample_path))
rep_full = LocusRepertoire(clonotypes=clonotypes, locus='TRB')
rep = downsample(rep_full, 20_000, random_seed=42)
print(f'Loaded: {rep_full.clonotype_count:,} clonotypes')
print(f'Downsampled to {rep.clonotype_count:,} clonotypes @ 20k reads')
/Users/mikesh/vcs/mirpy/.venv/lib/python3.12/site-packages/tqdm/auto.py:21: TqdmWarning: IProgress not found. Please update jupyter and ipywidgets. See https://ipywidgets.readthedocs.io/en/stable/user_install.html
from .autonotebook import tqdm as notebook_tqdm
Loaded: 1,150,027 clonotypes
Downsampled to 17,147 clonotypes @ 20k reads
[3]:
# Build edit-distance metaclonotypes using the high-level cluster_metaclonotypes API
# MetaclonotypeClusterConfig holds all method-specific parameters
cfg = MetaclonotypeClusterConfig(
method='edit_distance',
metric='hamming',
threshold=1,
graph_algo='components',
min_cluster_size=2,
n_jobs=4,
)
t0 = time.perf_counter()
meta = cluster_metaclonotypes(rep, cfg)
elapsed = time.perf_counter() - t0
print(f'Metaclonotypes: {meta.n_clusters:,} clusters (elapsed: {elapsed:.1f}s)')
print(f'Clustering rate: {meta.n_clusters / rep.clonotype_count:.2%} of clonotypes in a cluster')
# summarize_metaclonotypes: returns a Polars DataFrame with cluster-level stats
summary = summarize_metaclonotypes(rep, meta)
print('\nTop 5 metaclonotypes by duplicate_count:')
print(summary.sort('duplicate_count', descending=True).head(5))
Skipping 387 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 387 sequences with non-canonical amino acids (*, _, or non-standard chars)
Metaclonotypes: 666 clusters (elapsed: 5.7s)
Clustering rate: 3.88% of clonotypes in a cluster
Top 5 metaclonotypes by duplicate_count:
shape: (5, 8)
┌────────────┬───────────┬────────────┬───────────┬────────────┬───────────┬───────────┬───────────┐
│ cluster_id ┆ n_members ┆ duplicate_ ┆ umi_count ┆ representa ┆ represent ┆ represent ┆ represent │
│ --- ┆ --- ┆ count ┆ --- ┆ tive_clono ┆ ative_jun ┆ ative_v_g ┆ ative_j_g │
│ str ┆ u32 ┆ --- ┆ i64 ┆ type_id ┆ ction_aa ┆ ene ┆ ene │
│ ┆ ┆ i64 ┆ ┆ --- ┆ --- ┆ --- ┆ --- │
│ ┆ ┆ ┆ ┆ str ┆ str ┆ str ┆ str │
╞════════════╪═══════════╪════════════╪═══════════╪════════════╪═══════════╪═══════════╪═══════════╡
│ 0 ┆ 4 ┆ 632 ┆ 0 ┆ null ┆ null ┆ null ┆ null │
│ 1 ┆ 2 ┆ 314 ┆ 0 ┆ null ┆ null ┆ null ┆ null │
│ 166 ┆ 214 ┆ 228 ┆ 0 ┆ null ┆ null ┆ null ┆ null │
│ 20 ┆ 186 ┆ 204 ┆ 0 ┆ null ┆ null ┆ null ┆ null │
│ 373 ┆ 144 ┆ 146 ┆ 0 ┆ null ┆ null ┆ null ┆ null │
└────────────┴───────────┴────────────┴───────────┴────────────┴───────────┴───────────┴───────────┘
[4]:
# Functional diversity: compare clonotypic vs metaclonotype-level metrics
div_clono = summarize_clonotypes(rep.clonotypes)
div_func = functional_diversity(rep, meta)
print('Clonotypic diversity:')
print(f' abundance={div_clono.abundance:,} diversity={div_clono.diversity:,}')
print(f' shannon={div_clono.shannon:.3f} gini_simpson={div_clono.gini_simpson:.4f}')
print(f' chao1={div_clono.chao1:.1f}')
print('\nFunctional diversity (metaclonotype level):')
print(f' abundance={div_func.abundance:,} diversity={div_func.diversity:,}')
print(f' shannon={div_func.shannon:.3f} gini_simpson={div_func.gini_simpson:.4f}')
print(f' chao1={div_func.chao1:.1f}')
print(f'\nFunctional / clonotypic Shannon ratio: {div_func.shannon / div_clono.shannon:.3f}')
print(f'Compression (clusters / clonotypes): {div_func.diversity / div_clono.diversity:.4f}')
Clonotypic diversity:
abundance=20,000 diversity=17,147
shannon=9.295 gini_simpson=0.9983
chao1=349810.9
Functional diversity (metaclonotype level):
abundance=4,135 diversity=666
shannon=4.874 gini_simpson=0.9603
chao1=666.0
Functional / clonotypic Shannon ratio: 0.524
Compression (clusters / clonotypes): 0.0388
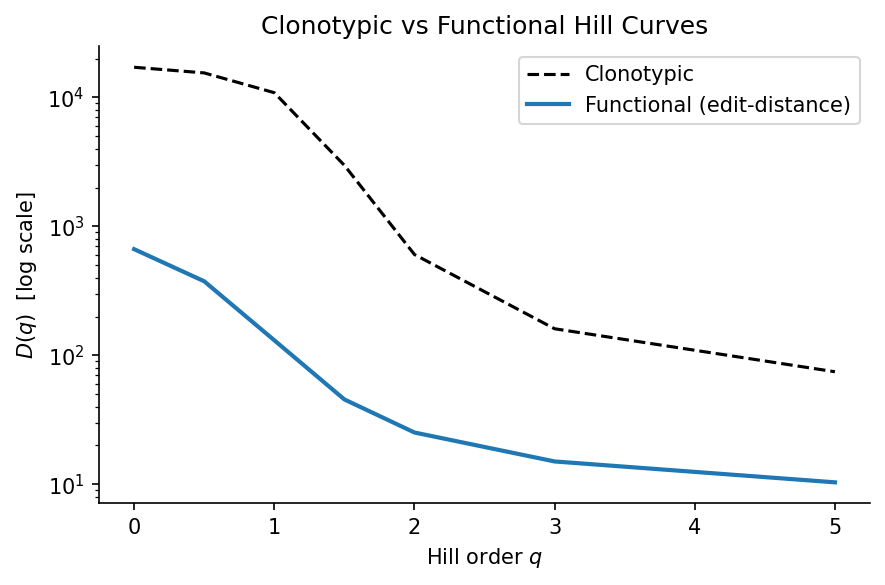
Functional Hill curves#
The Hill diversity curve \(D(q)\) at order \(q\):
\(q=0\): species richness
\(q=1\): Shannon entropy (effective number of species)
\(q=2\): inverse Simpson (dominated by abundant species)
Functional curves summarize how diversity changes across orders at the metaclonotype level.
[5]:
# Hill curves: clonotypic vs functional (edit-distance metaclonotypes)
q_values = [0.0, 0.5, 1.0, 1.5, 2.0, 3.0, 5.0]
hill_clono = hill_curve_clonotypes(rep.clonotypes, q_values=q_values)
hill_func = functional_hill_curve(rep, meta, q_values=q_values)
fig, ax = plt.subplots(figsize=(6, 4))
ax.plot(hill_clono['q'], hill_clono['hill'], 'k--', lw=1.5, label='Clonotypic')
ax.plot(hill_func['q'], hill_func['hill'], 'C0', lw=2, label='Functional (edit-distance)')
ax.set_yscale('log')
ax.set_xlabel('Hill order $q$')
ax.set_ylabel('$D(q)$ [log scale]')
ax.set_title('Clonotypic vs Functional Hill Curves')
ax.legend()
plt.tight_layout()
plt.show()
Leiden graph clustering#
graph_algo='leiden' merges multi-hop dense neighbourhoods; it finds fewer but larger clusters than the 'components' algorithm. Use Leiden when you want biologically coherent groupings that bridge closely related but not directly linked CDR3s.
[6]:
# Compare components vs Leiden graph algorithm
results = {}
for algo in ('components', 'leiden'):
cfg_algo = MetaclonotypeClusterConfig(
method='edit_distance',
metric='hamming',
threshold=1,
graph_algo=algo,
min_cluster_size=2,
n_jobs=4,
)
m = cluster_metaclonotypes(rep, cfg_algo)
d = functional_diversity(rep, m)
results[algo] = {'n_clusters': m.n_clusters, 'shannon': d.shannon, 'gini_simpson': d.gini_simpson}
print(f'{algo:12s}: clusters={m.n_clusters:,} shannon={d.shannon:.3f} gini_simpson={d.gini_simpson:.4f}')
print(f'\nLeiden / components cluster ratio: {results["leiden"]["n_clusters"] / results["components"]["n_clusters"]:.3f}')
Skipping 387 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 387 sequences with non-canonical amino acids (*, _, or non-standard chars)
components : clusters=666 shannon=4.874 gini_simpson=0.9603
Skipping 387 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 387 sequences with non-canonical amino acids (*, _, or non-standard chars)
leiden : clusters=677 shannon=5.085 gini_simpson=0.9657
Leiden / components cluster ratio: 1.017
ALICE-based metaclonotypes#
ALICE detects statistically over-represented clonotypes (expanded beyond Pgen expectation). add_alice_metadata runs the ALICE enrichment and stores q-values in each clonotype’s clone_metadata. MetaclonotypeClusterConfig(method='alice') then wraps enriched clonotypes and their 1-mismatch neighbours into metaclonotypes.
[7]:
# Run ALICE enrichment, then build metaclonotypes from significant hits
rep_alice = add_alice_metadata(rep, species='human', metric='hamming', match_mode='vj', n_jobs=4)
cfg_alice = MetaclonotypeClusterConfig(method='alice', q_value_max=0.05)
meta_alice = cluster_metaclonotypes(rep_alice, cfg_alice)
div_alice = functional_diversity(rep_alice, meta_alice)
print(f'ALICE metaclonotypes: {meta_alice.n_clusters:,} clusters')
print(f'Functional Shannon: {div_alice.shannon:.3f}')
print(f'Functional Chao1: {div_alice.chao1:.1f}')
print(f'\nTop ALICE clusters:')
print(summarize_metaclonotypes(rep_alice, meta_alice).sort('duplicate_count', descending=True).head(5))
Skipping 2 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 3 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 13 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 3 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 3 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 3 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 10 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 3 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 3 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 3 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 13 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 5 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 3 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 3 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 10 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 3 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 5 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 9 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 7 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 3 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 4 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 3 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 3 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 3 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 3 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 3 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 6 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 11 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 3 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 16 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 7 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 4 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 4 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 4 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 6 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 4 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 4 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 9 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 4 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 2 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
Skipping 1 sequences with non-canonical amino acids (*, _, or non-standard chars)
ALICE metaclonotypes: 855 clusters
Functional Shannon: 5.136
Functional Chao1: 855.1
Top ALICE clusters:
shape: (5, 8)
┌────────────┬───────────┬────────────┬───────────┬────────────┬───────────┬───────────┬───────────┐
│ cluster_id ┆ n_members ┆ duplicate_ ┆ umi_count ┆ representa ┆ represent ┆ represent ┆ represent │
│ --- ┆ --- ┆ count ┆ --- ┆ tive_clono ┆ ative_jun ┆ ative_v_g ┆ ative_j_g │
│ str ┆ u32 ┆ --- ┆ i64 ┆ type_id ┆ ction_aa ┆ ene ┆ ene │
│ ┆ ┆ i64 ┆ ┆ --- ┆ --- ┆ --- ┆ --- │
│ ┆ ┆ ┆ ┆ str ┆ str ┆ str ┆ str │
╞════════════╪═══════════╪════════════╪═══════════╪════════════╪═══════════╪═══════════╪═══════════╡
│ alice_mc_0 ┆ 3 ┆ 631 ┆ 0 ┆ 0 ┆ CATATSGEH ┆ TRBV27*01 ┆ TRBJ2-3*0 │
│ ┆ ┆ ┆ ┆ ┆ TDTQYF ┆ ┆ 1 │
│ alice_mc_5 ┆ 2 ┆ 630 ┆ 0 ┆ 477498 ┆ CAIATSGEH ┆ TRBV27*01 ┆ TRBJ2-3*0 │
│ 47 ┆ ┆ ┆ ┆ ┆ TDTQYF ┆ ┆ 1 │
│ alice_mc_2 ┆ 2 ┆ 9 ┆ 0 ┆ 40 ┆ CASSLNQGS ┆ TRBV12-4* ┆ TRBJ1-6*0 │
│ ┆ ┆ ┆ ┆ ┆ PLHF ┆ 01 ┆ 1 │
│ alice_mc_3 ┆ 2 ┆ 9 ┆ 0 ┆ 672 ┆ CASSLNQGS ┆ TRBV12-4* ┆ TRBJ1-6*0 │
│ 8 ┆ ┆ ┆ ┆ ┆ PLHF ┆ 01 ┆ 1 │
│ alice_mc_1 ┆ 3 ┆ 8 ┆ 0 ┆ 26 ┆ CSAPDSTDT ┆ TRBV20-1* ┆ TRBJ2-3*0 │
│ ┆ ┆ ┆ ┆ ┆ QYF ┆ 01 ┆ 1 │
└────────────┴───────────┴────────────┴───────────┴────────────┴───────────┴───────────┴───────────┘
Method comparison summary#
Method |
Config |
Clusters |
Notes |
|---|---|---|---|
|
|
small, tight |
Exact 1-mismatch |
|
|
fewer, larger |
Merges dense neighbourhoods |
|
|
enriched seeds |
Statistically significant only |
For a full method comparison including TCRdist, TCREmp, and GLIPH, see metaclonotype_method_compare.ipynb.