ALICE Analysis: YFV19, Ankylosing Spondylitis (AS), and MLR TCR-seq Datasets#
This notebook runs a MIR implementation of ALICE (Antigen-specific Lymphocyte Identification by Clustering of Expanded sequences) on three benchmark datasets from isalgo/airr_benchmark. The implementation is inspired by ideas described in the ALICE paper and is not a literal line-by-line reimplementation of the original software.
YF (Yellow Fever vaccine) — 6 donors × 2 time points (day 0, day 15), TRB repertoires.
AS (Ankylosing Spondylitis) — 4 donors, synovial fluid CD8+ TRB repertoires.
MLR (Mixed Lymphocyte Reaction) — 24 samples (fresh vs proliferating) from Adaptive immunoSEQ.
Reference: Pogorelyy MV, Minervina AA, Touzel MP, et al. PLoS Biol. 2019;17(6):e3000314. doi:10.1371/journal.pbio.3000314. PMID:31194732.
Runtime note: Uses N_JOBS=8 for parallel OLGA Pgen computation and trie-based neighbourhood search. pgen_mode="1mm" (paper-correct) is used throughout; exact pgen pre-screen skips sequences with n ≤ N×pgen_exact so only a small fraction of eligible sequences incur the expensive 1mm OLGA call.
[1]:
"""Cell 1: Environment setup, imports, and dataset download."""
import gzip
import sys
import random
import time
import resource
import warnings
from pathlib import Path
from collections import defaultdict
import importlib.metadata
import matplotlib.patches as mpatches
import matplotlib.pyplot as plt
import numpy as np
import pandas as pd
import polars as pl
from IPython.display import display
from scipy.stats import mannwhitneyu
# Environment versions for reproducibility
print(f"Python {sys.version.split()[0]}")
for _pkg in ["mirpy-lib", "polars", "numpy", "pandas", "matplotlib", "scipy"]:
try:
print(f" {_pkg}: {importlib.metadata.version(_pkg)}")
except importlib.metadata.PackageNotFoundError:
pass
repo_root = (
Path.cwd().resolve().parent
if Path.cwd().name == "notebooks"
else Path.cwd().resolve()
)
if str(repo_root) not in sys.path:
sys.path.insert(0, str(repo_root))
from mir.biomarkers.alice import compute_alice, alice_hit_clusters
from mir.common.clonotype import Clonotype
from mir.common.filter import filter_functional
from mir.common.parser import AdaptiveParser, ClonotypeTableParser
from mir.common.repertoire import LocusRepertoire, infer_locus
from mir.graph.edit_distance_graph import build_edit_distance_graph
from mir.utils.notebook_assets import ensure_airr_benchmark, ensure_airr_benchmark_alice
SEED = 42
np.random.seed(SEED)
N_JOBS = 8
# BH-FDR threshold from Pogorelyy et al. 2019 (PLoS Biol): 0.001
FDR_THRESH = 0.001
# n_neighbors counts self, so N_MIN_NEIGHBORS=2 means self + ≥1 Hamming-1 neighbour,
# matching the neighbourhood enrichment criterion from Pogorelyy et al. 2019.
N_MIN_NEIGHBORS = 2
MLR_FRESH_CAP = 300_000 # cap fresh MLR samples to keep neighbourhood search tractable
# Pgen mode — all three compute λ = N × Σ pgen(Hamming-1 neighbourhood):
# "mc" — MC pool of 10M synthetic seqs (paper used 1000M; increase MC_POOL_N if
# RAM allows, ~2 GB for 10M, ~20 GB for 100M). Pool is disk-cached via
# ControlManager on first run; subsequent runs load from disk (fast).
# "1mm" — OLGA analytical 1mm pgen (~70 ms/seq). Exact but slow for >100K clonotypes.
# "exact" — OLGA exact pgen only. Underestimates λ → inflates hit counts; NOT correct.
ALICE_PGEN_MODE = "mc"
# Pool size: original ALICE paper used 1_000_000_000. 100M is the safe default on 32 GB machines.
MC_POOL_N = 10_000_000
RAM_LIMIT_GB = 28.0 # YF+MLR reach ~26 GB peak with 10M hash pool; hardware limit 32 GB
def _rss_gb() -> float:
return resource.getrusage(resource.RUSAGE_SELF).ru_maxrss / 1e9
def _check_ram(label: str = "") -> None:
rss = _rss_gb()
tag = f"[{label}] " if label else ""
flag = " *** OVER LIMIT ***" if rss > RAM_LIMIT_GB else ""
print(f" {tag}RSS {rss:.2f} GB{flag}")
plt.rcParams.update({
"font.family": "sans-serif",
"font.sans-serif": ["Arial", "Helvetica", "DejaVu Sans"],
"font.size": 10,
"axes.titlesize": 11,
"axes.labelsize": 10,
"xtick.labelsize": 9,
"ytick.labelsize": 9,
"legend.fontsize": 9,
"figure.dpi": 150,
"savefig.dpi": 300,
"axes.linewidth": 0.8,
"axes.spines.top": False,
"axes.spines.right": False,
"xtick.major.size": 3.5,
"ytick.major.size": 3.5,
"xtick.direction": "out",
"ytick.direction": "out",
"axes.grid": False,
"figure.figsize": (7, 5),
})
warnings.filterwarnings("ignore", category=FutureWarning)
warnings.filterwarnings("ignore", category=DeprecationWarning)
BENCHMARK_DIR_YF_AS = ensure_airr_benchmark_alice(repo_root=repo_root, subsets=["yf", "as"])
YF_DIR = BENCHMARK_DIR_YF_AS / "alice" / "yf"
AS_DIR = BENCHMARK_DIR_YF_AS / "alice" / "as"
BENCHMARK_DIR_MLR = ensure_airr_benchmark(repo_root=repo_root, allow_patterns=["alice/mlr/*"])
MLR_DIR = BENCHMARK_DIR_MLR / "alice" / "mlr"
VDJDB_PATH = BENCHMARK_DIR_YF_AS / "vdjdb" / "vdjdb-2025-12-29" / "vdjdb.slim.txt.gz"
print(f"\nYF_DIR = {YF_DIR} (exists: {YF_DIR.exists()})")
print(f"AS_DIR = {AS_DIR} (exists: {AS_DIR.exists()})")
print(f"MLR_DIR = {MLR_DIR} (exists: {MLR_DIR.exists()})")
print(f"\nConfig: N_JOBS={N_JOBS}, ALICE_PGEN_MODE={ALICE_PGEN_MODE!r}, MC_POOL_N={MC_POOL_N:,}, "
f"FDR={FDR_THRESH}, RAM_LIMIT={RAM_LIMIT_GB} GB")
_check_ram("after import")
_t0_notebook = time.perf_counter()
_peak_rss: dict[str, float] = {}
# ─── ALICE cluster / convergent-response analysis helpers ─────────────────────
# alice_hit_clusters is imported from mir.biomarkers.alice (V-gene restricted,
# non_enriched_neighbors option available).
def cluster_stats(hits_with_ids: pd.DataFrame) -> dict:
"""Summarise cluster-size distribution for one sample's ALICE hits."""
if hits_with_ids.empty:
return {"n_hits": 0, "n_clusters": 0, "max_size": 0,
"median_size": 0.0, "n_multi": 0}
sizes = hits_with_ids.groupby("cluster_id").size()
return {
"n_hits": len(hits_with_ids),
"n_clusters": len(sizes),
"max_size": int(sizes.max()),
"median_size": float(sizes.median()),
"n_multi": int((sizes > 1).sum()),
}
def find_shared_hits(tables: dict, min_samples: int = 2) -> pd.DataFrame:
"""Return ALICE hit CDR3s present in >= min_samples samples (exact CDR3 match).
Args:
tables: Mapping of sample-key → DataFrame with a ``junction_aa`` column.
min_samples: Minimum number of samples a CDR3 must appear in.
Returns:
DataFrame with columns ``junction_aa``, ``n_samples``, ``samples``.
"""
rows = [
{"key": str(k), "junction_aa": cdr3}
for k, tbl in tables.items()
for cdr3 in tbl["junction_aa"].unique()
if not tbl.empty
]
if not rows:
return pd.DataFrame(columns=["junction_aa", "n_samples", "samples"])
df = pd.DataFrame(rows)
g = df.groupby("junction_aa")["key"].agg(list).reset_index()
g["n_samples"] = g["key"].apply(len)
g.rename(columns={"key": "samples"}, inplace=True)
return (
g[g["n_samples"] >= min_samples]
.sort_values("n_samples", ascending=False)
.reset_index(drop=True)
)
Python 3.12.12
mirpy-lib: 1.1.0
polars: 1.40.1
numpy: 1.26.4
pandas: 3.0.3
matplotlib: 3.10.9
scipy: 1.17.1
/Users/mikesh/vcs/mirpy/.venv/lib/python3.12/site-packages/tqdm/auto.py:21: TqdmWarning: IProgress not found. Please update jupyter and ipywidgets. See https://ipywidgets.readthedocs.io/en/stable/user_install.html
from .autonotebook import tqdm as notebook_tqdm
YF_DIR = /Users/mikesh/vcs/mirpy/notebooks/assets/large/airr_benchmark/alice/yf (exists: True)
AS_DIR = /Users/mikesh/vcs/mirpy/notebooks/assets/large/airr_benchmark/alice/as (exists: True)
MLR_DIR = /Users/mikesh/vcs/mirpy/notebooks/assets/large/airr_benchmark/alice/mlr (exists: True)
Config: N_JOBS=8, ALICE_PGEN_MODE='mc', MC_POOL_N=10,000,000, FDR=0.001, RAM_LIMIT=28.0 GB
[after import] RSS 0.32 GB
Yellow Fever (YF) Dataset#
Parse YF samples#
Files are named (e.g. , ). auto-detects column names and infers locus from the prefix.
[2]:
"""Cell 2: Parse YF samples from MiXCR TSV.gz files."""
parser_yf = ClonotypeTableParser()
warnings.filterwarnings("ignore", category=FutureWarning)
def _parse_yf_filename(path: Path) -> tuple[str, int]:
"""Return (donor, day) from `<donor>_d<day>.tsv.gz`."""
stem = path.name.replace(".tsv.gz", "").replace(".tsv", "")
donor, day_part = stem.split("_d")
return donor, int(day_part)
yf_samples: dict[tuple[str, int], LocusRepertoire] = {}
for fp in sorted(YF_DIR.glob("*.tsv.gz")):
try:
donor, day = _parse_yf_filename(fp)
except ValueError:
print(f" Skipping unexpected filename: {fp.name}")
continue
clonotypes = parser_yf.parse(str(fp))
trb = [c for c in clonotypes if c.locus == "TRB"]
rep = filter_functional(LocusRepertoire(clonotypes=trb, locus="TRB", repertoire_id=fp.stem))
yf_samples[(donor, day)] = rep
print(f" {fp.name}: {rep.clonotype_count:,} TRB clonotypes")
print(f"\nLoaded {len(yf_samples)} YF samples")
P1_d0.tsv.gz: 604,303 TRB clonotypes
P1_d15.tsv.gz: 930,938 TRB clonotypes
P2_d0.tsv.gz: 634,418 TRB clonotypes
P2_d15.tsv.gz: 1,218,195 TRB clonotypes
Q1_d0.tsv.gz: 371,649 TRB clonotypes
Q1_d15.tsv.gz: 456,935 TRB clonotypes
Q2_d0.tsv.gz: 706,531 TRB clonotypes
Q2_d15.tsv.gz: 623,617 TRB clonotypes
S1_d0.tsv.gz: 550,606 TRB clonotypes
S1_d15.tsv.gz: 688,621 TRB clonotypes
S2_d0.tsv.gz: 731,419 TRB clonotypes
S2_d15.tsv.gz: 848,550 TRB clonotypes
Loaded 12 YF samples
[3]:
"""Cell 2b: Dataset size diagnostic after parsing YF."""
_all_yf_count = sum(rep.clonotype_count for rep in yf_samples.values())
_all_yf_unique = len(set(
c.junction_aa for rep in yf_samples.values() for c in rep.clonotypes
))
print(f"YF: {_all_yf_count:,} total clonotypes, {_all_yf_unique:,} unique CDR3s across {len(yf_samples)} samples")
print(f" Pgen ({ALICE_PGEN_MODE!r}) computed only for sequences with n_neighbors >= {N_MIN_NEIGHBORS}.")
_check_ram("YF parsed")
YF: 8,365,782 total clonotypes, 6,025,751 unique CDR3s across 12 samples
Pgen ('mc') computed only for sequences with n_neighbors >= 2.
[YF parsed] RSS 6.96 GB
Run ALICE on YF samples#
ALICE scores each clonotype by comparing its Hamming-1 neighbourhood size n against the Poisson expectation λ = N × pgen_1mm, where N is the number of clonotypes sharing the same V+J gene pair (match_mode="vj") and pgen_1mm is the sum of OLGA Pgen over all CDR3s within Hamming-1 of the query (Pogorelyy, Minervina, Shugay et al. 2019). Benjamini–Hochberg FDR is applied per sample.
ALICE hit criterion: n_neighbors ≥ 2 (self + ≥1 real Hamming-1 neighbour) AND q_value < 0.001 (BH-FDR). Isolated sequences with Pgen ≪ 1/N are Pgen-significant but not convergent — the neighbourhood filter removes them.
Cluster analysis: after ALICE, hits are connected into clusters by 1mm edges among hits only (not expanded into the full repertoire). Convergent hits shared across multiple samples identify antigen-driven public responses.
[4]:
"""Cell 3: Run ALICE on YF samples; collect cluster members."""
yf_alice_tables: dict[tuple[str, int], pd.DataFrame] = {}
yf_alice_rows = []
_t0_yf = time.perf_counter()
print(f"YF ALICE start — pgen_mode={ALICE_PGEN_MODE!r}, n_jobs={N_JOBS}")
_check_ram("start")
for (donor, day), rep in sorted(yf_samples.items()):
_ts = time.perf_counter()
with warnings.catch_warnings():
warnings.simplefilter("ignore")
result = compute_alice(
rep,
species="human",
match_mode="vj",
pgen_mode=ALICE_PGEN_MODE,
min_neighbors=N_MIN_NEIGHBORS,
mc_n_pool=MC_POOL_N,
as_table=True,
n_jobs=N_JOBS,
)
_te = time.perf_counter()
tbl = result.table.to_pandas()
yf_alice_tables[(donor, day)] = tbl
hits = tbl[(tbl["n_neighbors"] >= N_MIN_NEIGHBORS) & (tbl["q_value"] < FDR_THRESH)].copy()
yf_alice_rows.append({
"Donor": donor,
"Day": day,
"Clonotypes": rep.clonotype_count,
"ALICE clusters": len(hits),
"Cluster %": 100 * len(hits) / max(1, rep.clonotype_count),
"wall_s": round(_te - _ts, 1),
})
print(
f" {donor} day{day:>3}: {rep.clonotype_count:,} clonotypes → {len(hits)} ALICE members"
f" [{_te - _ts:.1f}s, RSS {_rss_gb():.2f} GB]"
)
_yf_wall = time.perf_counter() - _t0_yf
print(f"\nYF ALICE done: {_yf_wall:.1f}s total | RSS {_rss_gb():.2f} GB")
_check_ram("end")
df_yf_summary = pd.DataFrame(yf_alice_rows).sort_values(["Donor", "Day"]).reset_index(drop=True)
display(
df_yf_summary.drop(columns=["wall_s"]).style
.format({"Cluster %": "{:.1f}%", "Clonotypes": "{:,}", "ALICE clusters": "{:,}"})
.background_gradient(subset=["ALICE clusters", "Cluster %"], cmap="Blues")
.set_caption(
f"YF — ALICE cluster members per sample "
f"(n_neighbors ≥ {N_MIN_NEIGHBORS}, BH-FDR < {FDR_THRESH})"
)
.set_table_styles([{"selector": "caption", "props": [("font-weight", "bold"), ("font-size", "10pt")]}])
)
_peak_rss["yf"] = _rss_gb()
YF ALICE start — pgen_mode='mc', n_jobs=8
[start] RSS 6.96 GB
P1 day 0: 604,303 clonotypes → 45709 ALICE members [74.4s, RSS 9.02 GB]
P1 day 15: 930,938 clonotypes → 91278 ALICE members [63.7s, RSS 11.18 GB]
P2 day 0: 634,418 clonotypes → 52259 ALICE members [36.5s, RSS 11.79 GB]
P2 day 15: 1,218,195 clonotypes → 135544 ALICE members [88.9s, RSS 13.74 GB]
Q1 day 0: 371,649 clonotypes → 19912 ALICE members [20.7s, RSS 13.74 GB]
Q1 day 15: 456,935 clonotypes → 29344 ALICE members [25.8s, RSS 13.80 GB]
Q2 day 0: 706,531 clonotypes → 57489 ALICE members [43.0s, RSS 14.78 GB]
Q2 day 15: 623,617 clonotypes → 51720 ALICE members [40.2s, RSS 15.43 GB]
S1 day 0: 550,606 clonotypes → 35835 ALICE members [29.8s, RSS 15.87 GB]
S1 day 15: 688,621 clonotypes → 50809 ALICE members [38.8s, RSS 15.87 GB]
S2 day 0: 731,419 clonotypes → 58149 ALICE members [43.7s, RSS 16.23 GB]
S2 day 15: 848,550 clonotypes → 88114 ALICE members [54.7s, RSS 16.38 GB]
YF ALICE done: 560.5s total | RSS 16.38 GB
[end] RSS 16.38 GB
| Donor | Day | Clonotypes | ALICE clusters | Cluster % | |
|---|---|---|---|---|---|
| 0 | P1 | 0 | 604,303 | 45,709 | 7.6% |
| 1 | P1 | 15 | 930,938 | 91,278 | 9.8% |
| 2 | P2 | 0 | 634,418 | 52,259 | 8.2% |
| 3 | P2 | 15 | 1,218,195 | 135,544 | 11.1% |
| 4 | Q1 | 0 | 371,649 | 19,912 | 5.4% |
| 5 | Q1 | 15 | 456,935 | 29,344 | 6.4% |
| 6 | Q2 | 0 | 706,531 | 57,489 | 8.1% |
| 7 | Q2 | 15 | 623,617 | 51,720 | 8.3% |
| 8 | S1 | 0 | 550,606 | 35,835 | 6.5% |
| 9 | S1 | 15 | 688,621 | 50,809 | 7.4% |
| 10 | S2 | 0 | 731,419 | 58,149 | 8.0% |
| 11 | S2 | 15 | 848,550 | 88,114 | 10.4% |
[5]:
"""Cell 3b: YF — cluster connectivity and convergent-hit analysis."""
# ── Per-sample: cluster hits into connected components (1mm among hits only) ──
yf_cluster_rows = []
yf_hits_clustered: dict[tuple, pd.DataFrame] = {}
for (donor, day), tbl in sorted(yf_alice_tables.items()):
hits = tbl[(tbl["n_neighbors"] >= N_MIN_NEIGHBORS) & (tbl["q_value"] < FDR_THRESH)].copy()
if not hits.empty:
hits = alice_hit_clusters(hits)
yf_hits_clustered[(donor, day)] = hits
stats = cluster_stats(hits)
yf_cluster_rows.append({"Donor": donor, "Day": day, **stats})
df_yf_clusters = (
pd.DataFrame(yf_cluster_rows)
.sort_values(["Donor", "Day"])
.reset_index(drop=True)
)
display(
df_yf_clusters.style
.format({"median_size": "{:.1f}"})
.background_gradient(subset=["n_clusters", "max_size", "n_multi"], cmap="Blues")
.set_caption(
f"YF — ALICE cluster connectivity per sample (FDR < {FDR_THRESH})\n"
"n_clusters = connected components | n_multi = clusters with ≥2 hits"
)
.set_table_styles([{"selector": "caption", "props": [("font-weight", "bold"), ("font-size", "10pt")]}])
)
# ── Cross-sample: hits shared across any ≥2 YF samples ────────────────────────
yf_shared_any = find_shared_hits(
{f"{d}_d{dy}": t for (d, dy), t in yf_hits_clustered.items()},
min_samples=2,
)
print(f"\nYF shared ALICE hits in ≥2 samples: {len(yf_shared_any)}")
# ── Day-15 convergent hits (present as ALICE hit in ≥2 day15 donors) ──────────
yf_d15_tables = {
d: t for (d, dy), t in yf_hits_clustered.items() if dy == 15 and not t.empty
}
yf_d15_shared = find_shared_hits(yf_d15_tables, min_samples=2)
print(f"YF day15 convergent hits (≥2 day15 donors): {len(yf_d15_shared)}")
if not yf_d15_shared.empty:
display(
yf_d15_shared.head(20).style
.background_gradient(subset=["n_samples"], cmap="Blues")
.set_caption("YF — convergent ALICE hits at day 15 (shared across ≥2 donors)")
.set_table_styles([{"selector": "caption", "props": [("font-weight", "bold"), ("font-size", "10pt")]}])
)
# ── Fold-change check: day15 clusters > day0 ──────────────────────────────────
print("\nDay0 vs Day15 — cluster counts per donor:")
for donor in sorted(df_yf_clusters["Donor"].unique()):
d0_row = df_yf_clusters[(df_yf_clusters["Donor"] == donor) & (df_yf_clusters["Day"] == 0)]
d15_row = df_yf_clusters[(df_yf_clusters["Donor"] == donor) & (df_yf_clusters["Day"] == 15)]
if len(d0_row) and len(d15_row):
h0, h15 = d0_row["n_hits"].values[0], d15_row["n_hits"].values[0]
c0, c15 = d0_row["n_clusters"].values[0], d15_row["n_clusters"].values[0]
ok = "✓" if h15 > h0 else "⚠"
print(f" {donor}: hits {h0}→{h15} ({h15/max(h0,1):.1f}×) "
f"clusters {c0}→{c15} ({c15/max(c0,1):.1f}×) {ok}")
| Donor | Day | n_hits | n_clusters | max_size | median_size | n_multi | |
|---|---|---|---|---|---|---|---|
| 0 | P1 | 0 | 45709 | 10033 | 1087 | 2.0 | 5084 |
| 1 | P1 | 15 | 91278 | 24683 | 3621 | 1.0 | 11943 |
| 2 | P2 | 0 | 52259 | 11341 | 1721 | 2.0 | 5805 |
| 3 | P2 | 15 | 135544 | 31489 | 4492 | 1.0 | 15173 |
| 4 | Q1 | 0 | 19912 | 6018 | 340 | 2.0 | 3031 |
| 5 | Q1 | 15 | 29344 | 7686 | 708 | 2.0 | 3906 |
| 6 | Q2 | 0 | 57489 | 15340 | 1379 | 1.0 | 7434 |
| 7 | Q2 | 15 | 51720 | 14951 | 1488 | 1.0 | 7317 |
| 8 | S1 | 0 | 35835 | 9244 | 1414 | 1.0 | 4393 |
| 9 | S1 | 15 | 50809 | 12648 | 2086 | 1.0 | 6103 |
| 10 | S2 | 0 | 58149 | 15673 | 1861 | 1.0 | 7424 |
| 11 | S2 | 15 | 88114 | 19018 | 4464 | 1.0 | 9171 |
YF shared ALICE hits in ≥2 samples: 69978
YF day15 convergent hits (≥2 day15 donors): 35476
| junction_aa | samples | n_samples | |
|---|---|---|---|
| 0 | CSARGGSYNEQFF | ['P1', 'P2', 'Q1', 'Q2', 'S1', 'S2'] | 6 |
| 1 | CASSLSGGSYNEQFF | ['P1', 'P2', 'Q1', 'Q2', 'S1', 'S2'] | 6 |
| 2 | CSARDSGSSYEQYF | ['P1', 'P2', 'Q1', 'Q2', 'S1', 'S2'] | 6 |
| 3 | CSARTSGTDTQYF | ['P1', 'P2', 'Q1', 'Q2', 'S1', 'S2'] | 6 |
| 4 | CSVDRGTEAFF | ['P1', 'P2', 'Q1', 'Q2', 'S1', 'S2'] | 6 |
| 5 | CSVDRGTDTQYF | ['P1', 'P2', 'Q1', 'Q2', 'S1', 'S2'] | 6 |
| 6 | CSARTSGSYNEQFF | ['P1', 'P2', 'Q1', 'Q2', 'S1', 'S2'] | 6 |
| 7 | CSARTSGSYEQYF | ['P1', 'P2', 'Q1', 'Q2', 'S1', 'S2'] | 6 |
| 8 | CASSLSGGTEAFF | ['P1', 'P2', 'Q1', 'Q2', 'S1', 'S2'] | 6 |
| 9 | CSARGQGDTEAFF | ['P1', 'P2', 'Q1', 'Q2', 'S1', 'S2'] | 6 |
| 10 | CASSYSRGTEAFF | ['P1', 'P2', 'Q1', 'Q2', 'S1', 'S2'] | 6 |
| 11 | CSVDRGNTGELFF | ['P1', 'P2', 'Q1', 'Q2', 'S1', 'S2'] | 6 |
| 12 | CSVDRGNTEAFF | ['P1', 'P2', 'Q1', 'Q2', 'S1', 'S2'] | 6 |
| 13 | CASSLSGNEQFF | ['P1', 'P2', 'Q1', 'Q2', 'S1', 'S2'] | 6 |
| 14 | CASSLSGNTEAFF | ['P1', 'P2', 'Q1', 'Q2', 'S1', 'S2'] | 6 |
| 15 | CASSLSGNTGELFF | ['P1', 'P2', 'Q1', 'Q2', 'S1', 'S2'] | 6 |
| 16 | CSVDRNTEAFF | ['P1', 'P2', 'Q1', 'Q2', 'S1', 'S2'] | 6 |
| 17 | CASSLSGANVLTF | ['P1', 'P2', 'Q1', 'Q2', 'S1', 'S2'] | 6 |
| 18 | CASSLSDTQYF | ['P1', 'P2', 'Q1', 'Q2', 'S1', 'S2'] | 6 |
| 19 | CASSYGGGTEAFF | ['P1', 'P2', 'Q1', 'Q2', 'S1', 'S2'] | 6 |
Day0 vs Day15 — cluster counts per donor:
P1: hits 45709→91278 (2.0×) clusters 10033→24683 (2.5×) ✓
P2: hits 52259→135544 (2.6×) clusters 11341→31489 (2.8×) ✓
Q1: hits 19912→29344 (1.5×) clusters 6018→7686 (1.3×) ✓
Q2: hits 57489→51720 (0.9×) clusters 15340→14951 (1.0×) ⚠
S1: hits 35835→50809 (1.4×) clusters 9244→12648 (1.4×) ✓
S2: hits 58149→88114 (1.5×) clusters 15673→19018 (1.2×) ✓
YF Visualization#
Paired bar chart comparing ALICE cluster member fraction (%) at day 0 and day 15 per donor. A day-15 increase is expected for HLA-A*02 donors responding to LLWNGPMAV.
[6]:
"""Cell 4: YF — ALICE hits per donor, Fig 2a style.
Left panel: total ALICE hits (count). Right panel: YF-specific (VDJdb LLWNGPMAV).
"""
import gzip as _gzip
import polars as _pl
# ── Load VDJdb LLWNGPMAV reference ────────────────────────────────────────────
with _gzip.open(VDJDB_PATH, "rt") as _fh:
_vdb = _pl.read_csv(_fh, separator="\t", infer_schema_length=10000, ignore_errors=True)
llw_cdr3s: set[str] = set(
_vdb.filter(
(_pl.col("antigen.epitope") == "LLWNGPMAV") &
(_pl.col("gene") == "TRB") &
(_pl.col("species") == "HomoSapiens")
).select("cdr3").unique("cdr3")["cdr3"].to_list()
)
print(f"VDJdb LLWNGPMAV TRB reference: {len(llw_cdr3s)} unique CDR3s")
llw_rows = []
for (donor, day), tbl in sorted(yf_alice_tables.items()):
hits = tbl[(tbl["n_neighbors"] >= N_MIN_NEIGHBORS) & (tbl["q_value"] < FDR_THRESH)]
n_llw = int(hits["junction_aa"].isin(llw_cdr3s).sum()) if not hits.empty else 0
llw_rows.append({"Donor": donor, "Day": day, "LLW hits": n_llw})
df_llw = pd.DataFrame(llw_rows).sort_values(["Donor", "Day"]).reset_index(drop=True)
# ── Donor color palette (matches paper Fig 2a) ────────────────────────────────
YF_DONOR_COLORS = {
"P1": "#4292c6",
"P2": "#d94701",
"Q1": "#fd8d3c",
"Q2": "#74c476",
"S1": "#2ca25f",
"S2": "#006d2c",
}
donors_ordered = sorted(df_yf_summary["Donor"].unique())
days_ordered = sorted(df_yf_summary["Day"].unique())
n_don = len(donors_ordered)
bw = 0.11
gap = 0.28
def _yf_barplot(ax, df_pivot_col, ylabel, title):
for d_i, donor in enumerate(donors_ordered):
for t_i, day in enumerate(days_ordered):
row = df_pivot_col[(df_pivot_col["Donor"] == donor) & (df_pivot_col["Day"] == day)]
val = float(row[df_pivot_col.columns[-1]].values[0]) if len(row) else 0.0
x = t_i * (n_don * bw + gap) + d_i * bw
ax.bar(x, val, bw * 0.92,
color=YF_DONOR_COLORS.get(donor, "#aaaaaa"),
label=donor if t_i == 0 else "_",
edgecolor="white", linewidth=0.25, alpha=0.92)
tick_x = [t_i * (n_don * bw + gap) + (n_don - 1) * bw / 2 for t_i in range(len(days_ordered))]
ax.set_xticks(tick_x)
ax.set_xticklabels([f"d{d}" for d in days_ordered])
ax.set_ylabel(ylabel)
ax.set_title(title, fontsize=10)
ax.spines[["top", "right"]].set_visible(False)
df_total = df_yf_summary[["Donor", "Day", "ALICE clusters"]].copy()
df_yf_llw = df_llw.copy()
fig, (ax1, ax2) = plt.subplots(1, 2, figsize=(11, 4.5))
_yf_barplot(ax1, df_total, "ALICE hits", "Total")
_yf_barplot(ax2, df_yf_llw, "ALICE hits", "YF specific")
handles = [mpatches.Patch(facecolor=YF_DONOR_COLORS[d], edgecolor="none", label=d)
for d in donors_ordered]
ax2.legend(handles=handles, title="donor", frameon=False, loc="upper left",
fontsize=8, title_fontsize=8)
plt.suptitle("YF vaccine response — ALICE hits per donor\n(Pogorelyy, Minervina, Shugay et al. 2019 Fig 2a)",
fontsize=10, y=1.01)
plt.tight_layout()
plt.show()
d15_total = df_llw[df_llw["Day"] == 15]["LLW hits"].sum()
d0_total = df_llw[df_llw["Day"] == 0]["LLW hits"].sum()
print(f"LLW hits: day15={d15_total}, day0={d0_total}")
if d15_total > d0_total:
print("✓ Day-15 LLW hits > day-0 (expected for YFV response)")
else:
print("⚠ WARNING: day-15 LLW hits do NOT exceed day-0")
VDJdb LLWNGPMAV TRB reference: 409 unique CDR3s
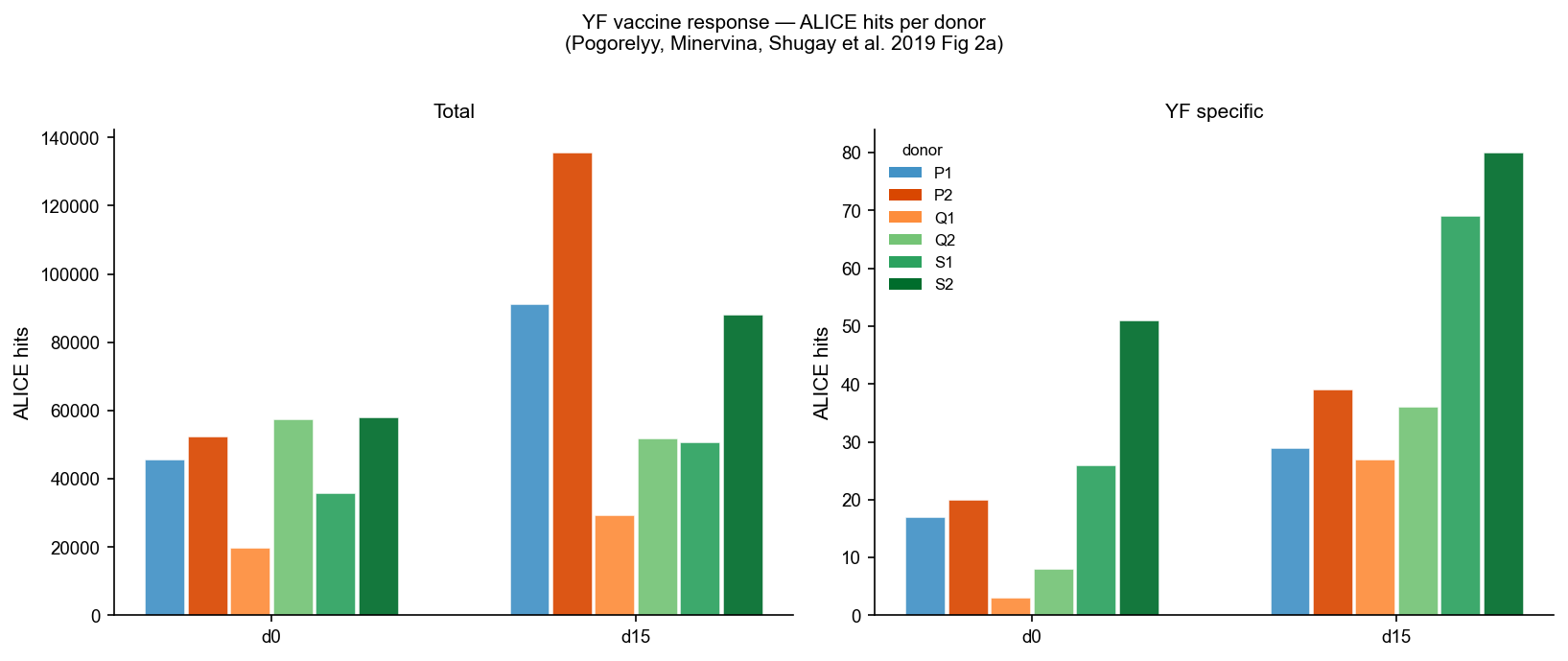
LLW hits: day15=280, day0=125
✓ Day-15 LLW hits > day-0 (expected for YFV response)
VDJdb LLWNGPMAV overlap#
ALICE hits are intersected with VDJdb 2025-12-29 TRB entries for LLWNGPMAV (all HLA-A*02 entries). Exact CDR3 matching is used — the YFV-specific T cells in this dataset were sorted from the same donors, so exact identity is the appropriate criterion. Donors carrying HLA-A*02 are expected to show a stronger day-15 signal because this epitope is immunodominant in YFV-vaccinated A*02 individuals (Pogorelyy, Minervina, Shugay et al. 2019).
[7]:
"""Cell 5: VDJdb LLWNGPMAV table (computed in Cell 4)."""
display(
df_llw.style
.format({"LLW hits": "{:,}"})
.background_gradient(subset=["LLW hits"], cmap="Blues")
.set_caption("ALICE hits matching LLWNGPMAV (VDJdb 2025-12-29, TRB, exact match)")
.set_table_styles([{"selector": "caption", "props": [("font-weight", "bold"), ("font-size", "10pt")]}])
)
yf_d15_llw = df_llw[df_llw["Day"] == 15]["LLW hits"].sum()
yf_d0_llw = df_llw[df_llw["Day"] == 0]["LLW hits"].sum()
print(f"Day-15 LLW-matching ALICE hits: {yf_d15_llw}")
print(f"Day-0 LLW-matching ALICE hits: {yf_d0_llw}")
| Donor | Day | LLW hits | |
|---|---|---|---|
| 0 | P1 | 0 | 17 |
| 1 | P1 | 15 | 29 |
| 2 | P2 | 0 | 20 |
| 3 | P2 | 15 | 39 |
| 4 | Q1 | 0 | 3 |
| 5 | Q1 | 15 | 27 |
| 6 | Q2 | 0 | 8 |
| 7 | Q2 | 15 | 36 |
| 8 | S1 | 0 | 26 |
| 9 | S1 | 15 | 69 |
| 10 | S2 | 0 | 51 |
| 11 | S2 | 15 | 80 |
Day-15 LLW-matching ALICE hits: 280
Day-0 LLW-matching ALICE hits: 125
Ankylosing Spondylitis (AS) Dataset#
Parse AS samples#
Four donors from synovial fluid CD8+ TRB repertoires. Donor B27 status metadata is hard-coded from study design.
[8]:
"""Cell 6: Parse AS samples."""
AS_DONOR_META = {
1: "B27_pos",
2: "B27_pos",
3: "B27_neg",
4: "B27_pos",
}
as_samples: dict[int, LocusRepertoire] = {}
for donor_id, b27 in sorted(AS_DONOR_META.items()):
pattern = f"Donor{donor_id}_B27_{b27}_SF_CD8.tsv.gz"
fp = AS_DIR / pattern
if not fp.exists():
# Try globbing in case of slight filename variation.
candidates = list(AS_DIR.glob(f"Donor{donor_id}_*.tsv.gz"))
if not candidates:
print(f" WARNING: no file for Donor {donor_id} — skipping")
continue
fp = candidates[0]
clonotypes = parser_yf.parse(str(fp))
trb = [c for c in clonotypes if c.locus == "TRB"]
rep = filter_functional(LocusRepertoire(clonotypes=trb, locus="TRB", repertoire_id=fp.stem))
as_samples[donor_id] = rep
print(f" Donor {donor_id} ({b27}): {rep.clonotype_count:,} TRB clonotypes [{fp.name}]")
print(f"\nLoaded {len(as_samples)} AS samples")
Donor 1 (B27_pos): 29,193 TRB clonotypes [Donor1_B27_pos_SF_CD8.tsv.gz]
Donor 2 (B27_pos): 9,571 TRB clonotypes [Donor2_B27_pos_SF_CD8.tsv.gz]
Donor 3 (B27_neg): 38,404 TRB clonotypes [Donor3_B27_neg_SF_CD8.tsv.gz]
Donor 4 (B27_pos): 27,145 TRB clonotypes [Donor4_B27_pos_SF_CD8.tsv.gz]
Loaded 4 AS samples
[9]:
"""Cell 6b: Dataset size diagnostic after parsing AS."""
_all_as_count = sum(rep.clonotype_count for rep in as_samples.values())
_all_as_unique = len(set(
c.junction_aa for rep in as_samples.values() for c in rep.clonotypes
))
print(f"AS: {_all_as_count:,} total clonotypes, {_all_as_unique:,} unique CDR3s across {len(as_samples)} samples")
print(f" Pgen ({ALICE_PGEN_MODE!r}) computed only for sequences with n_neighbors >= {N_MIN_NEIGHBORS}.")
_check_ram("AS parsed")
AS: 104,313 total clonotypes, 100,453 unique CDR3s across 4 samples
Pgen ('mc') computed only for sequences with n_neighbors >= 2.
[AS parsed] RSS 16.48 GB
Run ALICE on AS samples#
Same parameters as YF (match_mode="vj", pgen_mode="1mm", BH-FDR < 0.001). B27+ donors (1, 2, 4) are expected to share convergent CDR3 neighbourhoods absent in the B27- donor (3) — driven by shared HLA-B27-restricted antigen responses in synovial tissue.
Cluster analysis identifies hits shared across B27+ donors as evidence for a public alloresponse.
[10]:
"""Cell 7: Run ALICE on AS samples; collect cluster members."""
as_alice_hits: dict[int, pd.DataFrame] = {}
as_alice_tables: dict[int, pd.DataFrame] = {}
as_alice_rows = []
_t0_as = time.perf_counter()
print(f"AS ALICE start — pgen_mode={ALICE_PGEN_MODE!r}, n_jobs={N_JOBS}")
_check_ram("start")
for donor_id, rep in sorted(as_samples.items()):
b27 = AS_DONOR_META[donor_id]
_ts = time.perf_counter()
with warnings.catch_warnings():
warnings.simplefilter("ignore")
result = compute_alice(
rep,
species="human",
match_mode="vj",
pgen_mode=ALICE_PGEN_MODE,
min_neighbors=N_MIN_NEIGHBORS,
mc_n_pool=MC_POOL_N,
as_table=True,
n_jobs=N_JOBS,
)
_te = time.perf_counter()
tbl = result.table.to_pandas()
hits = tbl[(tbl["n_neighbors"] >= N_MIN_NEIGHBORS) & (tbl["q_value"] < FDR_THRESH)].copy()
hits["donor_id"] = donor_id
hits["b27"] = b27
as_alice_hits[donor_id] = hits
as_alice_tables[donor_id] = tbl
as_alice_rows.append({
"Donor": donor_id,
"B27 status": b27,
"Clonotypes": rep.clonotype_count,
"ALICE clusters": len(hits),
"Cluster %": 100 * len(hits) / max(1, rep.clonotype_count),
"wall_s": round(_te - _ts, 1),
})
print(
f" Donor {donor_id} ({b27}): {rep.clonotype_count:,} clonotypes → {len(hits)} ALICE members"
f" [{_te - _ts:.1f}s, RSS {_rss_gb():.2f} GB]"
)
_as_wall = time.perf_counter() - _t0_as
print(f"\nAS ALICE done: {_as_wall:.1f}s total | RSS {_rss_gb():.2f} GB")
_check_ram("end")
df_as_summary = pd.DataFrame(as_alice_rows)
display(
df_as_summary.drop(columns=["wall_s"]).style
.format({"Cluster %": "{:.1f}%", "Clonotypes": "{:,}", "ALICE clusters": "{:,}"})
.background_gradient(subset=["Cluster %"], cmap="Blues")
.set_caption(
f"AS — ALICE cluster members per donor "
f"(n_neighbors ≥ {N_MIN_NEIGHBORS}, BH-FDR < {FDR_THRESH})"
)
.set_table_styles([{"selector": "caption", "props": [("font-weight", "bold"), ("font-size", "10pt")]}])
)
df_as_all_hits = pd.concat(as_alice_hits.values(), ignore_index=True)
print(f"\nTotal AS ALICE cluster members: {len(df_as_all_hits)}")
print(f"Unique junction_aa: {df_as_all_hits['junction_aa'].nunique()}")
_peak_rss["as"] = _rss_gb()
AS ALICE start — pgen_mode='mc', n_jobs=8
[start] RSS 16.48 GB
Donor 1 (B27_pos): 29,193 clonotypes → 1094 ALICE members [7.3s, RSS 16.48 GB]
Donor 2 (B27_pos): 9,571 clonotypes → 458 ALICE members [3.8s, RSS 16.48 GB]
Donor 3 (B27_neg): 38,404 clonotypes → 1413 ALICE members [7.7s, RSS 16.48 GB]
Donor 4 (B27_pos): 27,145 clonotypes → 1469 ALICE members [6.2s, RSS 16.48 GB]
AS ALICE done: 25.0s total | RSS 16.48 GB
[end] RSS 16.48 GB
| Donor | B27 status | Clonotypes | ALICE clusters | Cluster % | |
|---|---|---|---|---|---|
| 0 | 1 | B27_pos | 29,193 | 1,094 | 3.7% |
| 1 | 2 | B27_pos | 9,571 | 458 | 4.8% |
| 2 | 3 | B27_neg | 38,404 | 1,413 | 3.7% |
| 3 | 4 | B27_pos | 27,145 | 1,469 | 5.4% |
Total AS ALICE cluster members: 4434
Unique junction_aa: 3450
[11]:
"""Cell 7b: AS — cluster connectivity, joint B27+ analysis, and B27- exclusivity check.
Cluster algorithm: alice_hit_clusters() from mir.biomarkers.alice.
Uses V-gene-restricted 1mm CDR3 edges — only sequences sharing the same V-gene
family are connected, preventing spurious transitive merges across unrelated families.
Key result (Pogorelyy, Minervina, Shugay et al. 2019 Fig 2d): the motif CASSVGL[YF]STDTQYF (TRBV9 TRBJ2-3)
appears as the LARGEST connected component in pooled B27+ hits and is absent in B27-.
"""
# ── Per-donor: cluster hits into connected components (V-gene restricted) ─────
as_cluster_rows = []
as_hits_clustered: dict[int, pd.DataFrame] = {}
for donor_id, hits in sorted(as_alice_hits.items()):
b27 = AS_DONOR_META[donor_id]
clust = alice_hit_clusters(hits.copy()) if not hits.empty else hits
as_hits_clustered[donor_id] = clust
stats = cluster_stats(clust)
as_cluster_rows.append({"Donor": donor_id, "B27": b27, **stats})
df_as_clusters = pd.DataFrame(as_cluster_rows).sort_values("Donor").reset_index(drop=True)
display(
df_as_clusters.style
.format({"median_size": "{:.1f}"})
.background_gradient(subset=["n_clusters", "max_size", "n_multi"], cmap="Blues")
.set_caption(
f"AS — ALICE cluster connectivity per donor (FDR < {FDR_THRESH}, V-gene restricted)\n"
"n_clusters = connected components | n_multi = clusters with ≥2 hits"
)
.set_table_styles([{"selector": "caption", "props": [("font-weight", "bold"), ("font-size", "10pt")]}])
)
# ── Joint B27+ cluster analysis ────────────────────────────────────────────────
# Pool all B27+ ALICE hits (deduplicated by junction_aa + v_gene per donor) and
# cluster into connected components using V-gene-restricted 1mm edges.
# Expected: the CASSVGL[YF]STDTQYF (TRBV9 TRBJ2-3) motif is the largest cluster.
B27_POS_IDS = [d for d, status in AS_DONOR_META.items() if status == "B27_pos"]
B27_NEG_IDS = [d for d, status in AS_DONOR_META.items() if status == "B27_neg"]
# Deduplicate per donor before pooling to avoid inflated cluster sizes from
# multiple nucleotide sequences encoding the same amino acid.
b27_pos_hits_dedup = pd.concat(
[
as_alice_hits[d].drop_duplicates(subset=["junction_aa", "v_gene"]).assign(donor_id=d)
for d in B27_POS_IDS if d in as_alice_hits and not as_alice_hits[d].empty
],
ignore_index=True,
)
MOTIF_CDR3S = {
"CASSVGLYSTDTQYF", "CASSVGLFSTDTQYF", "CASSVGVYSTDTQYF",
"CASSVATYSTDTQYF", "CASSLGLFSTDTQYF", "CASSAGLFSTDTQYF",
"CASSPGLFSTDTQYF", "CASSVGGFGDTQYF",
}
print(f"\nJoint B27+ cluster analysis ({len(b27_pos_hits_dedup)} deduplicated hits from {len(B27_POS_IDS)} donors)")
b27_pos_clustered = alice_hit_clusters(b27_pos_hits_dedup.copy())
sizes = b27_pos_clustered.groupby("cluster_id").size().sort_values(ascending=False)
print(f" {len(sizes)} clusters, top-5 sizes: {sizes.head(5).tolist()}")
# ── Motif cluster: should be rank 1 ──────────────────────────────────────────
motif_hits = b27_pos_clustered[b27_pos_clustered["junction_aa"].isin(MOTIF_CDR3S)]
if not motif_hits.empty:
motif_cid = motif_hits.iloc[0]["cluster_id"]
motif_cluster = b27_pos_clustered[b27_pos_clustered["cluster_id"] == motif_cid].copy()
motif_size = len(motif_cluster)
motif_rank = int((sizes > motif_size).sum()) + 1
donors_in_cluster = sorted(motif_cluster["donor_id"].unique().tolist())
print(f" Motif cluster (CASSVGL[YF]STDTQYF, TRBV9): size={motif_size}, "
f"rank={motif_rank}/{len(sizes)}, donors={donors_in_cluster}")
motif_display = (
motif_cluster[["donor_id", "junction_aa", "v_gene", "j_gene", "q_value"]]
.drop_duplicates(subset=["junction_aa", "v_gene"])
.sort_values("q_value")
.reset_index(drop=True)
.rename(columns={"donor_id": "Donor", "junction_aa": "CDR3", "v_gene": "V",
"j_gene": "J", "q_value": "q-value"})
)
motif_display["motif"] = motif_display["CDR3"].isin(MOTIF_CDR3S).map(
{True: "★ key", False: ""}
)
display(
motif_display.style
.format({"q-value": "{:.2e}"})
.map(lambda v: "font-weight: bold; color: #c0392b" if v == "★ key" else "",
subset=["motif"])
.background_gradient(subset=["q-value"], cmap="Blues_r")
.set_caption(
f"AS — joint B27+ motif cluster (rank {motif_rank}, size {motif_size})\n"
"CASSVGL[YF]STDTQYF (TRBV9 TRBJ2-3) — AS-associated public TCR"
)
.set_table_styles([{"selector": "caption", "props": [("font-weight", "bold"),
("font-size", "10pt")]}])
)
else:
print(" WARNING: motif sequences not found among B27+ ALICE hits")
# ── Largest non-motif cluster for comparison ─────────────────────────────────
print("\n Largest cluster overall:")
lid = sizes.index[0]
lg = b27_pos_clustered[b27_pos_clustered["cluster_id"] == lid]
lg_donors = sorted(lg["donor_id"].unique().tolist())
print(f" size={len(lg)}, donors={lg_donors}")
lg_dedup = lg.drop_duplicates(subset=["junction_aa","v_gene"])
print(f" Top-5 members (by q_value): " +
", ".join(f"{r['junction_aa']} ({r['v_gene'].split('*')[0]})"
for _, r in lg_dedup.sort_values("q_value").head(5).iterrows()))
# ── Convergent hits across B27+ donors ────────────────────────────────────────
b27_pos_tables_dedup = {
str(d): as_alice_hits[d].drop_duplicates(subset=["junction_aa"])
for d in B27_POS_IDS if d in as_alice_hits and not as_alice_hits[d].empty
}
as_b27pos_shared = find_shared_hits(b27_pos_tables_dedup, min_samples=2)
print(f"\nAS convergent hits in ≥2 B27+ donors: {len(as_b27pos_shared)}")
if not as_b27pos_shared.empty:
display(
as_b27pos_shared.head(20).style
.background_gradient(subset=["n_samples"], cmap="Blues")
.set_caption("AS — B27+ convergent ALICE hits (shared across ≥2 B27+ donors, exact CDR3)")
.set_table_styles([{"selector": "caption", "props": [("font-weight", "bold"),
("font-size", "10pt")]}])
)
# ── B27- exclusivity check ────────────────────────────────────────────────────
b27_neg_cdr3s = {
cdr3
for d in B27_NEG_IDS
for cdr3 in (as_alice_hits[d]["junction_aa"].unique() if d in as_alice_hits else [])
}
if not as_b27pos_shared.empty:
n_b27pos_only = (~as_b27pos_shared["junction_aa"].isin(b27_neg_cdr3s)).sum()
print(f" Of those, {n_b27pos_only} are absent from B27- donor hits (B27-specific)")
motif_in_b27neg = any(cdr3 in b27_neg_cdr3s for cdr3 in MOTIF_CDR3S)
print(f" Motif sequences in B27- ALICE hits: {motif_in_b27neg} (expected: False)")
| Donor | B27 | n_hits | n_clusters | max_size | median_size | n_multi | |
|---|---|---|---|---|---|---|---|
| 0 | 1 | B27_pos | 1094 | 446 | 19 | 2.0 | 261 |
| 1 | 2 | B27_pos | 458 | 225 | 11 | 2.0 | 135 |
| 2 | 3 | B27_neg | 1413 | 524 | 39 | 2.0 | 297 |
| 3 | 4 | B27_pos | 1469 | 679 | 38 | 2.0 | 345 |
Joint B27+ cluster analysis (2389 deduplicated hits from 3 donors)
1123 clusters, top-5 sizes: [41, 18, 17, 15, 14]
Motif cluster (CASSVGL[YF]STDTQYF, TRBV9): size=41, rank=1/1123, donors=[1, 2, 4]
| Donor | CDR3 | V | J | q-value | motif | |
|---|---|---|---|---|---|---|
| 0 | 4 | CASSVGWYSTDTQYF | TRBV9*01 | TRBJ2-3*01 | 3.03e-67 | |
| 1 | 4 | CASSVGVYSTDTQYF | TRBV9*01 | TRBJ2-3*01 | 3.77e-38 | ★ key |
| 2 | 4 | CASSVGWFSTDTQYF | TRBV9*01 | TRBJ2-3*01 | 7.16e-38 | |
| 3 | 4 | CASSPGLYSTDTQYF | TRBV9*01 | TRBJ2-3*01 | 2.50e-35 | |
| 4 | 4 | CASSLGLYSTDTQYF | TRBV9*01 | TRBJ2-3*01 | 8.32e-32 | |
| 5 | 4 | CASSVGRYSTDTQYF | TRBV9*01 | TRBJ2-3*01 | 1.54e-30 | |
| 6 | 4 | CASSGGLYSTDTQYF | TRBV9*01 | TRBJ2-3*01 | 1.96e-29 | |
| 7 | 4 | CASSSGLYSTDTQYF | TRBV9*01 | TRBJ2-3*01 | 1.96e-29 | |
| 8 | 4 | CASSVGQYSTDTQYF | TRBV9*01 | TRBJ2-3*01 | 2.38e-27 | |
| 9 | 4 | CASSVALYSTDTQYF | TRBV9*01 | TRBJ2-3*01 | 1.89e-24 | |
| 10 | 4 | CASSHGLFSTDTQYF | TRBV9*01 | TRBJ2-3*01 | 1.37e-23 | |
| 11 | 4 | CASSFGLFSTDTQYF | TRBV9*01 | TRBJ2-3*01 | 7.02e-23 | |
| 12 | 4 | CASSVGVFSTDTQYF | TRBV9*01 | TRBJ2-3*01 | 9.75e-22 | |
| 13 | 1 | CASSVGLFSTDTQYF | TRBV9*01 | TRBJ2-3*01 | 1.57e-19 | ★ key |
| 14 | 1 | CASSVGLYSTDTQYF | TRBV9*01 | TRBJ2-3*01 | 9.60e-17 | ★ key |
| 15 | 4 | CASNLGLYSTDTQYF | TRBV9*01 | TRBJ2-3*01 | 2.40e-16 | |
| 16 | 2 | CASSAGLFSTDTQYF | TRBV9*01 | TRBJ2-3*01 | 4.63e-16 | ★ key |
| 17 | 2 | CASSVAYYSTDTQYF | TRBV9*01 | TRBJ2-3*01 | 1.16e-15 | |
| 18 | 2 | CASSVATYSTDTQYF | TRBV9*01 | TRBJ2-3*01 | 2.77e-12 | ★ key |
| 19 | 1 | CASSAGLYSTDTQYF | TRBV9*01 | TRBJ2-3*01 | 1.13e-11 | |
| 20 | 2 | CASNAGLFSTDTQYF | TRBV9*01 | TRBJ2-3*01 | 1.44e-11 | |
| 21 | 4 | CASSVARYSTDTQYF | TRBV9*01 | TRBJ2-3*01 | 1.58e-09 | |
| 22 | 2 | CASSPGLFSTDTQYF | TRBV9*01 | TRBJ2-3*01 | 3.10e-09 | ★ key |
| 23 | 4 | CASSPGVYSTDTQYF | TRBV9*01 | TRBJ2-3*01 | 5.14e-09 | |
| 24 | 1 | CASSVGSYSTDTQYF | TRBV9*01 | TRBJ2-3*01 | 5.27e-09 | |
| 25 | 2 | CASSVVTYSTDTQYF | TRBV9*01 | TRBJ2-3*01 | 7.04e-09 | |
| 26 | 2 | CASSVGDYSTDTQYF | TRBV9*01 | TRBJ2-3*01 | 5.30e-08 | |
| 27 | 1 | CASSSGLFSTDTQYF | TRBV9*01 | TRBJ2-3*01 | 1.51e-07 | |
| 28 | 1 | CASSLGLFSTDTQYF | TRBV9*01 | TRBJ2-3*01 | 2.45e-06 | ★ key |
| 29 | 4 | CASSVASYSTDTQYF | TRBV9*01 | TRBJ2-3*01 | 3.47e-06 | |
| 30 | 1 | CASSVGTFSTDTQYF | TRBV9*01 | TRBJ2-3*01 | 5.16e-06 | |
| 31 | 4 | CASSAGTYSTDTQYF | TRBV9*01 | TRBJ2-3*01 | 7.12e-04 |
Largest cluster overall:
size=41, donors=[1, 2, 4]
Top-5 members (by q_value): CASSVGWYSTDTQYF (TRBV9), CASSVGVYSTDTQYF (TRBV9), CASSVGWFSTDTQYF (TRBV9), CASSPGLYSTDTQYF (TRBV9), CASSLGLYSTDTQYF (TRBV9)
AS convergent hits in ≥2 B27+ donors: 22
| junction_aa | samples | n_samples | |
|---|---|---|---|
| 0 | CASSVGLYSTDTQYF | ['1', '2', '4'] | 3 |
| 1 | CASSAGLYSTDTQYF | ['1', '2', '4'] | 3 |
| 2 | CASSLGLAGGPNTGELFF | ['1', '2', '4'] | 3 |
| 3 | CASSVGLFSTDTQYF | ['1', '2', '4'] | 3 |
| 4 | CASSYSMGSNTGELFF | ['1', '2'] | 2 |
| 5 | CSVVAGGPYEQYF | ['1', '4'] | 2 |
| 6 | CSARTGDTEAFF | ['1', '2'] | 2 |
| 7 | CSARRGDTEAFF | ['1', '2'] | 2 |
| 8 | CSAREVYEQYF | ['1', '4'] | 2 |
| 9 | CATSDLGQGVGNEQFF | ['2', '4'] | 2 |
| 10 | CATSDLGAGEGNEQFF | ['2', '4'] | 2 |
| 11 | CATGTSGTYNEQFF | ['2', '4'] | 2 |
| 12 | CAISERGPNYGYTF | ['1', '4'] | 2 |
| 13 | CASSYRLAGLNEQFF | ['1', '4'] | 2 |
| 14 | CASSAGLFSTDTQYF | ['2', '4'] | 2 |
| 15 | CASSQDPGYEQYF | ['1', '4'] | 2 |
| 16 | CASSQDPGHEQYF | ['1', '4'] | 2 |
| 17 | CASSPGLFSTDTQYF | ['2', '4'] | 2 |
| 18 | CASSLLAGGNNEQFF | ['1', '4'] | 2 |
| 19 | CASSLGLFSTDTQYF | ['1', '4'] | 2 |
Of those, 20 are absent from B27- donor hits (B27-specific)
Motif sequences in B27- ALICE hits: False (expected: False)
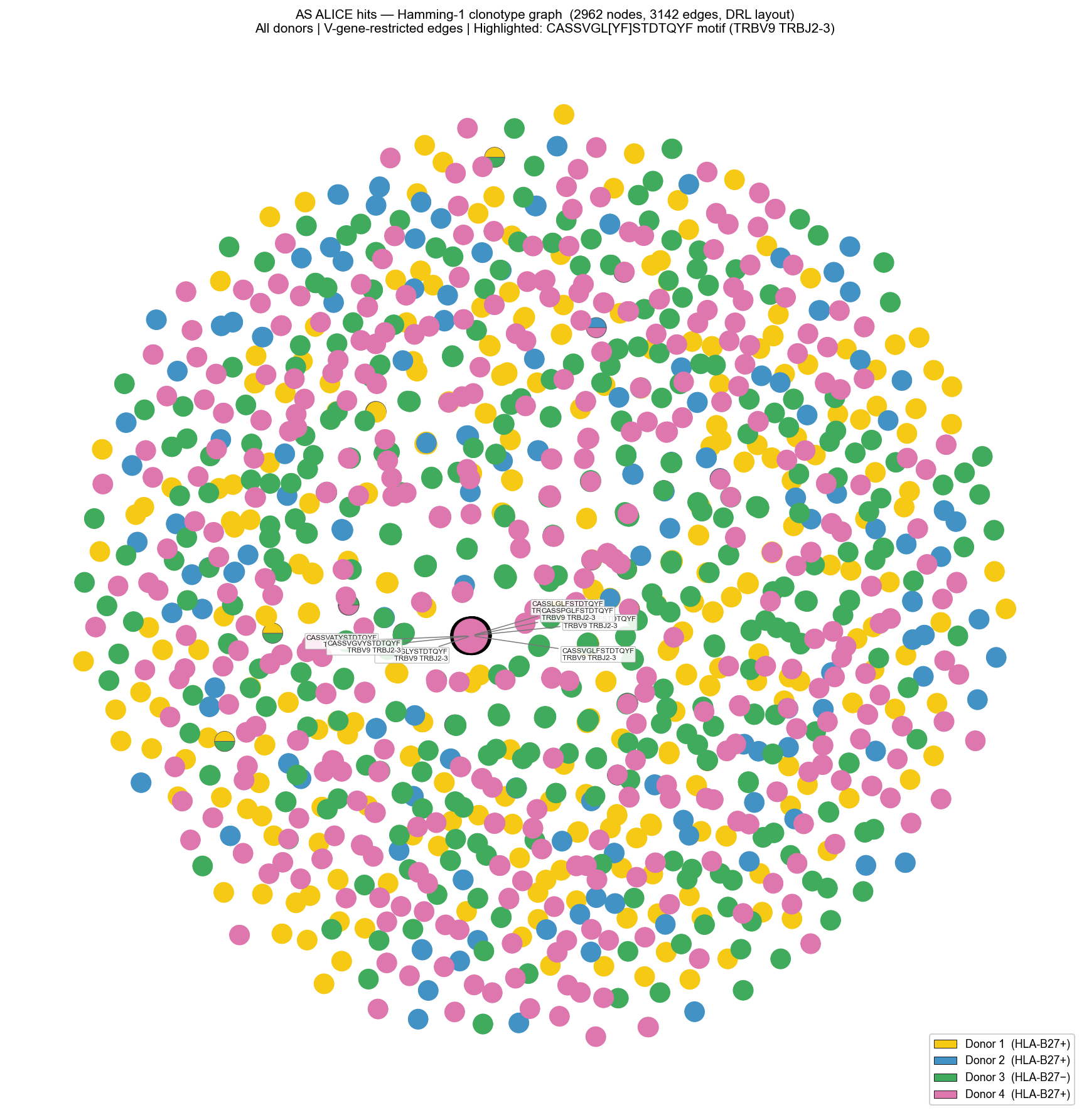
Build Hamming clonotype graph for AS ALICE hits#
Each ALICE hit clonotype becomes a graph node. Edges connect pairs with Hamming distance ≤ 1 in the same V-gene. We then build node color arrays for rendering with donor-specific colors.
[12]:
"""Cell 8: Build Hamming distance graph for AS ALICE hits — all donors.
Build from all-donor deduplicated hits so Donor 3 (B27_neg) is visible
alongside B27+ donors. Matches Pogorelyy, Minervina, Shugay et al. 2019 Fig 2d.
"""
# ── Donor color palette ────────────────────────────────────────────────────────
DONOR_COLORS = {
1: "#f6c915", # gold/yellow — B27_pos (Donor 1)
2: "#4292c6", # blue — B27_pos (Donor 2)
3: "#41ab5d", # green — B27_neg (Donor 3)
4: "#de77ae", # pink/rose — B27_pos (Donor 4)
}
# Highlight clonotypes of special interest (TRBV9 pair from literature).
HIGHLIGHT_CLONOTYPES = {
("TRBV9", "CASSVGLYSTDTQYF", "TRBJ2-3"),
("TRBV9", "CASSVGLFSTDTQYF", "TRBJ2-3"),
("TRBV9", "CASSVGVYSTDTQYF", "TRBJ2-3"),
("TRBV9", "CASSVATYSTDTQYF", "TRBJ2-3"),
("TRBV9", "CASSLGLFSTDTQYF", "TRBJ2-3"),
("TRBV9", "CASSAGLFSTDTQYF", "TRBJ2-3"),
("TRBV9", "CASSPGLFSTDTQYF", "TRBJ2-3"),
("TRBV9", "CASSVGGFGDTQYF", "TRBJ2-3"),
}
def _as_clonotype(row: pd.Series) -> Clonotype:
return Clonotype(
junction_aa=row.get("junction_aa") or "",
v_gene=row.get("v_gene") or "",
j_gene=row.get("j_gene") or "",
duplicate_count=1,
locus="TRB",
)
# Include ALL 4 donors (not just B27+) so Donor 3 (B27_neg) appears in the graph.
all_hits_dedup = pd.concat(
[
as_alice_hits[d].drop_duplicates(subset=["junction_aa", "v_gene"]).assign(donor_id=d)
for d in sorted(as_alice_hits)
if not as_alice_hits[d].empty
],
ignore_index=True,
)
clonotype_list = []
clonotype_donor_ids = []
for _, row in all_hits_dedup.iterrows():
clonotype_list.append(_as_clonotype(row))
clonotype_donor_ids.append(int(row["donor_id"]))
print(f"Graph input: {len(clonotype_list)} nodes ({len(as_alice_hits)} donors, all B27+/B27-)")
print(f"ALICE hit counts: {', '.join(f'Donor {d}: {len(as_alice_hits[d])}' for d in sorted(as_alice_hits))}")
g = build_edit_distance_graph(
clonotype_list,
metric="hamming",
threshold=1,
v_gene_match=True,
n_jobs=N_JOBS,
)
g.vs["donor_id"] = clonotype_donor_ids
print(f"Graph: {g.vcount()} nodes, {g.ecount()} edges")
Graph input: 3506 nodes (4 donors, all B27+/B27-)
ALICE hit counts: Donor 1: 1094, Donor 2: 458, Donor 3: 1413, Donor 4: 1469
Graph: 3506 nodes, 3142 edges
Render AS graph with donor colors and highlighted targets#
Rendering strategy:
Compute Fruchterman-Reingold layout
Draw edges (thin grey lines)
Single-donor nodes: filled circle with donor color
Multi-donor nodes: pie-chart wedges per donor
Star markers on target clonotypes
[13]:
"""Cell 9: Render AS Hamming graph — all donors, DRL layout, pie-chart shared nodes.
Matches Pogorelyy, Minervina, Shugay et al. 2019 Fig 2d.
Donor 3 (B27_neg) is shown in green; pie-chart nodes for sequences shared across donors.
"""
from matplotlib.patches import Circle, Wedge
# ── Subgraph: keep only connected components with ≥ 2 nodes ──────────────────
comp_sizes = g.connected_components().sizes()
subg = g
if max(comp_sizes) > 1:
large_comps = [i for i, sz in enumerate(comp_sizes) if sz >= 2]
keep = [v for v in range(g.vcount())
if g.connected_components().membership[v] in large_comps]
if keep:
subg = g.induced_subgraph(keep)
if subg.vcount() == 0:
print("No connected components with ≥ 2 nodes.")
else:
# DRL layout — Distributed Recursive Layout; separated clusters, cytoscape-like
np.random.seed(SEED)
layout = subg.layout("drl", seed=None)
coords = np.array(layout.coords)
mn, mx = coords.min(axis=0), coords.max(axis=0)
coords = (coords - mn) / np.where(mx - mn > 0, mx - mn, 1.0)
NODE_RADIUS = 0.011
HIGHLIGHT_RADIUS = NODE_RADIUS * 1.8
fig, ax = plt.subplots(figsize=(14, 12))
ax.set_aspect("equal")
ax.axis("off")
ax.set_xlim(-0.08, 1.08)
ax.set_ylim(-0.08, 1.08)
# Edges
for e in subg.es:
s, t = e.source, e.target
ax.plot([coords[s, 0], coords[t, 0]], [coords[s, 1], coords[t, 1]],
color="#cccccc", linewidth=0.4, zorder=1)
j_gene_in_graph = "j_gene" in subg.vs.attributes()
# Group vertices by (CDR3, v_gene) → merge same-sequence across donors for pie chart
key_to_nodes: dict[tuple, list[int]] = defaultdict(list)
for v in range(subg.vcount()):
key_to_nodes[(subg.vs[v]["name"], subg.vs[v]["v_gene"])].append(v)
highlight_positions: list[tuple[float, float, str]] = []
for key, node_ids in key_to_nodes.items():
donors_here = sorted({subg.vs[v]["donor_id"] for v in node_ids})
pos = coords[node_ids, :].mean(axis=0)
cx, cy = float(pos[0]), float(pos[1])
jaa = key[0]
vgene = key[1]
jgene = ""
if j_gene_in_graph:
raw = subg.vs[node_ids[0]]["j_gene"]
jgene = raw if raw is not None else ""
vg_stripped = vgene.split("*")[0]
jg_stripped = jgene.split("*")[0] if jgene else ""
is_highlight = (vg_stripped, jaa, jg_stripped) in HIGHLIGHT_CLONOTYPES
radius = HIGHLIGHT_RADIUS if is_highlight else NODE_RADIUS
if len(donors_here) == 1:
# Single donor — solid circle
ax.add_patch(Circle(
(cx, cy), radius,
facecolor=DONOR_COLORS.get(donors_here[0], "#cccccc"),
edgecolor="black" if is_highlight else "none",
linewidth=1.5 if is_highlight else 0, zorder=3))
else:
# Multiple donors — pie chart of donor colours
angle_per = 360.0 / len(donors_here)
for k, did in enumerate(donors_here):
ax.add_patch(Wedge(
(cx, cy), radius, k * angle_per, (k + 1) * angle_per,
facecolor=DONOR_COLORS.get(did, "#cccccc"),
edgecolor="black" if is_highlight else "#555555",
linewidth=1.5 if is_highlight else 0.4, zorder=3))
if is_highlight:
highlight_positions.append((cx, cy, jaa))
# Labels on highlighted (motif) nodes
if highlight_positions:
cx_c = float(np.mean([p[0] for p in highlight_positions]))
cy_c = float(np.mean([p[1] for p in highlight_positions]))
for cx, cy, label in highlight_positions:
dx, dy = cx - cx_c, cy - cy_c
norm_d = max(float(np.sqrt(dx**2 + dy**2)), 0.001)
dx, dy = dx / norm_d, dy / norm_d
offset = 0.10
tx = float(np.clip(cx + dx * offset, 0.03, 0.97))
ty = float(np.clip(cy + dy * offset, 0.03, 0.97))
ha = "left" if dx >= 0 else "right"
ax.annotate(
f"{label}\nTRBV9 TRBJ2-3",
xy=(cx, cy), xytext=(tx, ty),
fontsize=5.5, ha=ha, va="center", zorder=7, color="#222222",
arrowprops=dict(arrowstyle="-", color="#777777", lw=0.7),
bbox=dict(boxstyle="round,pad=0.15", facecolor="white",
alpha=0.88, edgecolor="#aaaaaa", linewidth=0.5))
# Legend
legend_handles = []
for did in sorted(DONOR_COLORS):
b27 = AS_DONOR_META[did]
hla = "HLA-B27+" if b27 == "B27_pos" else "HLA-B27−"
legend_handles.append(mpatches.Patch(
facecolor=DONOR_COLORS[did], edgecolor="black", linewidth=0.4,
label=f"Donor {did} ({hla})"))
ax.legend(handles=legend_handles, loc="lower right", fontsize=8.5,
frameon=True, framealpha=0.9, edgecolor="#cccccc")
ax.set_title(
f"AS ALICE hits — Hamming-1 clonotype graph "
f"({subg.vcount()} nodes, {subg.ecount()} edges, DRL layout)\n"
f"All donors | V-gene-restricted edges | "
f"Highlighted: CASSVGL[YF]STDTQYF motif (TRBV9 TRBJ2-3)",
fontsize=10)
plt.tight_layout()
# ── Consistency check ─────────────────────────────────────────────────────
comps = subg.connected_components()
j_gene_in_subg = "j_gene" in subg.vs.attributes()
sizes = sorted([(len(comps[ci]), ci) for ci in range(len(comps))], reverse=True)
largest_sz, largest_idx = sizes[0]
print(f"Components: {len(comps)}, sizes top-5: {[s for s, _ in sizes[:5]]}")
print(f"Highlighted nodes in graph: {len(highlight_positions)}/{len(HIGHLIGHT_CLONOTYPES)}")
motif_comp_idx = None
for ci in range(len(comps)):
for v in comps[ci]:
vg = (subg.vs[v]["v_gene"] or "").split("*")[0]
jaa = subg.vs[v]["name"] or ""
_jg = (subg.vs[v]["j_gene"] or "") if j_gene_in_subg else ""
jg = _jg.split("*")[0] if _jg else ""
if (vg, jaa, jg) in HIGHLIGHT_CLONOTYPES:
motif_comp_idx = ci
break
if motif_comp_idx is not None:
break
if motif_comp_idx is not None:
motif_nodes = list(comps[motif_comp_idx])
motif_rank = next(i+1 for i,(_, ci) in enumerate(sizes) if ci == motif_comp_idx)
motif_donors = sorted({subg.vs[v]["donor_id"] for v in motif_nodes})
n_hl = sum(
1 for v in motif_nodes
if ((subg.vs[v]["v_gene"] or "").split("*")[0],
subg.vs[v]["name"] or "",
((subg.vs[v]["j_gene"] or "").split("*")[0]
if j_gene_in_subg else "")) in HIGHLIGHT_CLONOTYPES)
print(f" Motif cluster: rank={motif_rank}/{len(comps)}, size={len(motif_nodes)}, donors={motif_donors}")
print(f" Highlighted in motif: {n_hl}")
if motif_rank == 1:
print(" ✓ Motif IS rank-1 (Pogorelyy, Minervina, Shugay et al. 2019 Fig 2d)")
else:
print(f" ⚠ Motif rank={motif_rank}")
else:
print(" ⚠ No highlight sequences found in graph")
plt.show()
Components: 991, sizes top-5: [41, 28, 27, 27, 20]
Highlighted nodes in graph: 7/8
Motif cluster: rank=1/991, size=41, donors=[1, 2, 4]
Highlighted in motif: 14
✓ Motif IS rank-1 (Pogorelyy, Minervina, Shugay et al. 2019 Fig 2d)
Mixed Lymphocyte Reaction (MLR) Dataset#
Load Adaptive files and build TRB repertoires#
The parser maps the Adaptive columns to AIRR-style fields. Files are parsed into single-locus TRB repertoires. Gene names are normalized (e.g. TCRBV01 → TRBV1).
[14]:
"""Cell 10: Parse MLR samples from Adaptive immunoSEQ files."""
parser_adaptive = AdaptiveParser(locus="TRB")
def parse_mlr_filename(path: Path) -> dict[str, object]:
"""Return sample metadata encoded in the MLR filename."""
stem = path.name.removesuffix(".tsv.gz").removesuffix(".tsv")
parts = stem.split("_")
if "Fresh" in parts:
condition = "fresh"
idx = parts.index("Fresh")
elif "Proliferating" in parts:
condition = "proliferating"
idx = parts.index("Proliferating")
else:
raise ValueError(f"Could not infer condition from {path.name}")
return {
"sample_id": stem,
"sample_group": "_".join(parts[:idx]),
"condition": condition,
"replicate": int(parts[-1]),
}
mlr_files = sorted(fp for fp in MLR_DIR.glob("*.tsv.gz") if "Stimulator" not in fp.name)
print(f"Found {len(mlr_files)} MLR files")
mlr_samples: dict[str, LocusRepertoire] = {}
mlr_rows: list[dict[str, object]] = []
for fp in mlr_files:
meta = parse_mlr_filename(fp)
rep = parser_adaptive.parse_file(fp, sample_id=str(meta["sample_id"]), locus="TRB")
rep_functional = rep.subsample_functional()
if meta["condition"] == "fresh" and rep_functional.clonotype_count > MLR_FRESH_CAP:
_sampled = random.sample(rep_functional.clonotypes, MLR_FRESH_CAP)
rep_functional = LocusRepertoire(
clonotypes=_sampled, locus="TRB",
repertoire_id=rep_functional.repertoire_id,
)
rep_functional.repertoire_metadata.update(meta)
mlr_samples[str(meta["sample_id"])] = rep_functional
mlr_rows.append({
**meta,
"n_clonotypes_total": rep.clonotype_count,
"n_clonotypes": rep_functional.clonotype_count,
"n_duplicates_total": rep.duplicate_count,
"n_duplicates": rep_functional.duplicate_count,
})
print(f" {fp.name}: {rep.clonotype_count:,} total → {rep_functional.clonotype_count:,} functional")
df_mlr_samples = pd.DataFrame(mlr_rows).sort_values(["sample_group", "condition", "replicate"]).reset_index(drop=True)
display(
df_mlr_samples.style
.format({"n_clonotypes_total": "{:,}", "n_clonotypes": "{:,}",
"n_duplicates_total": "{:,}", "n_duplicates": "{:,}"})
.set_caption("MLR samples — loaded functional clonotypes")
.set_table_styles([{"selector": "caption", "props": [("font-weight", "bold"), ("font-size", "10pt")]}])
)
Found 24 MLR files
MLR7_TCR1_Fresh_1.tsv.gz: 562,409 total → 300,000 functional
MLR7_TCR1_Fresh_2.tsv.gz: 448,953 total → 300,000 functional
MLR7_TCR1_Proliferating_1.tsv.gz: 45,698 total → 40,301 functional
MLR7_TCR1_Proliferating_2.tsv.gz: 45,971 total → 40,405 functional
MLR8_TCR2_Fresh_1.tsv.gz: 848,893 total → 300,000 functional
MLR8_TCR2_Fresh_2.tsv.gz: 945,294 total → 300,000 functional
MLR8_TCR2_Proliferating_1.tsv.gz: 104,389 total → 93,251 functional
MLR8_TCR2_Proliferating_2.tsv.gz: 88,868 total → 79,518 functional
MLR9_TCR3_Fresh_1.tsv.gz: 706,881 total → 300,000 functional
MLR9_TCR3_Fresh_2.tsv.gz: 578,431 total → 300,000 functional
MLR9_TCR3_Proliferating_1.tsv.gz: 76,291 total → 68,522 functional
MLR9_TCR3_Proliferating_2.tsv.gz: 68,886 total → 62,046 functional
TCR4_Fresh_Responder_R_1.tsv.gz: 180,520 total → 164,296 functional
TCR4_Fresh_Responder_R_2.tsv.gz: 243,485 total → 222,012 functional
TCR4_Proliferating_1.tsv.gz: 67,261 total → 60,915 functional
TCR4_Proliferating_2.tsv.gz: 67,610 total → 61,133 functional
TCR5_Fresh_Responder_R_1.tsv.gz: 706,253 total → 300,000 functional
TCR5_Fresh_Responder_R_2.tsv.gz: 627,904 total → 300,000 functional
TCR5_Proliferating_1.tsv.gz: 87,702 total → 78,893 functional
TCR5_Proliferating_2.tsv.gz: 68,662 total → 61,517 functional
TCR6_Fresh_Responder_R_1.tsv.gz: 654,314 total → 300,000 functional
TCR6_Fresh_Responder_R_2.tsv.gz: 721,281 total → 300,000 functional
TCR6_Proliferating_1.tsv.gz: 80,243 total → 73,229 functional
TCR6_Proliferating_2.tsv.gz: 84,066 total → 76,699 functional
| sample_id | sample_group | condition | replicate | n_clonotypes_total | n_clonotypes | n_duplicates_total | n_duplicates | |
|---|---|---|---|---|---|---|---|---|
| 0 | MLR7_TCR1_Fresh_1 | MLR7_TCR1 | fresh | 1 | 562,409 | 300,000 | 39,060,366 | 22,342,313 |
| 1 | MLR7_TCR1_Fresh_2 | MLR7_TCR1 | fresh | 2 | 448,953 | 300,000 | 14,774,134 | 10,201,749 |
| 2 | MLR7_TCR1_Proliferating_1 | MLR7_TCR1 | proliferating | 1 | 45,698 | 40,301 | 28,247,803 | 26,145,533 |
| 3 | MLR7_TCR1_Proliferating_2 | MLR7_TCR1 | proliferating | 2 | 45,971 | 40,405 | 31,989,495 | 29,412,778 |
| 4 | MLR8_TCR2_Fresh_1 | MLR8_TCR2 | fresh | 1 | 848,893 | 300,000 | 21,189,395 | 7,075,768 |
| 5 | MLR8_TCR2_Fresh_2 | MLR8_TCR2 | fresh | 2 | 945,294 | 300,000 | 32,165,682 | 10,517,176 |
| 6 | MLR8_TCR2_Proliferating_1 | MLR8_TCR2 | proliferating | 1 | 104,389 | 93,251 | 31,406,916 | 27,112,376 |
| 7 | MLR8_TCR2_Proliferating_2 | MLR8_TCR2 | proliferating | 2 | 88,868 | 79,518 | 17,719,903 | 15,213,872 |
| 8 | MLR9_TCR3_Fresh_1 | MLR9_TCR3 | fresh | 1 | 706,881 | 300,000 | 20,815,699 | 8,799,036 |
| 9 | MLR9_TCR3_Fresh_2 | MLR9_TCR3 | fresh | 2 | 578,431 | 300,000 | 17,854,323 | 8,890,743 |
| 10 | MLR9_TCR3_Proliferating_1 | MLR9_TCR3 | proliferating | 1 | 76,291 | 68,522 | 45,196,078 | 41,153,619 |
| 11 | MLR9_TCR3_Proliferating_2 | MLR9_TCR3 | proliferating | 2 | 68,886 | 62,046 | 27,912,916 | 25,488,967 |
| 12 | TCR4_Fresh_Responder_R_1 | TCR4 | fresh | 1 | 180,520 | 164,296 | 10,856,962 | 9,993,513 |
| 13 | TCR4_Fresh_Responder_R_2 | TCR4 | fresh | 2 | 243,485 | 222,012 | 8,971,185 | 8,252,747 |
| 14 | TCR4_Proliferating_1 | TCR4 | proliferating | 1 | 67,261 | 60,915 | 10,349,431 | 9,540,670 |
| 15 | TCR4_Proliferating_2 | TCR4 | proliferating | 2 | 67,610 | 61,133 | 12,136,264 | 11,174,355 |
| 16 | TCR5_Fresh_Responder_R_1 | TCR5 | fresh | 1 | 706,253 | 300,000 | 11,711,864 | 5,083,358 |
| 17 | TCR5_Fresh_Responder_R_2 | TCR5 | fresh | 2 | 627,904 | 300,000 | 12,114,878 | 5,477,289 |
| 18 | TCR5_Proliferating_1 | TCR5 | proliferating | 1 | 87,702 | 78,893 | 14,774,830 | 12,127,067 |
| 19 | TCR5_Proliferating_2 | TCR5 | proliferating | 2 | 68,662 | 61,517 | 14,822,695 | 12,226,119 |
| 20 | TCR6_Fresh_Responder_R_1 | TCR6 | fresh | 1 | 654,314 | 300,000 | 10,684,479 | 4,772,330 |
| 21 | TCR6_Fresh_Responder_R_2 | TCR6 | fresh | 2 | 721,281 | 300,000 | 10,568,646 | 4,178,273 |
| 22 | TCR6_Proliferating_1 | TCR6 | proliferating | 1 | 80,243 | 73,229 | 12,187,182 | 11,094,518 |
| 23 | TCR6_Proliferating_2 | TCR6 | proliferating | 2 | 84,066 | 76,699 | 13,724,578 | 12,489,332 |
[15]:
"""Cell 10b: Dataset size diagnostic after parsing MLR."""
_all_mlr_count = sum(rep.clonotype_count for rep in mlr_samples.values())
_all_mlr_unique = len(set(
c.junction_aa for rep in mlr_samples.values() for c in rep.clonotypes
))
print(f"MLR: {_all_mlr_count:,} total clonotypes, {_all_mlr_unique:,} unique CDR3s across {len(mlr_samples)} samples")
print(f" Fresh samples capped at {MLR_FRESH_CAP:,} clonotypes.")
print(f" Pgen ({ALICE_PGEN_MODE!r}) computed only for sequences with n_neighbors >= {N_MIN_NEIGHBORS}.")
_check_ram("MLR parsed")
MLR: 4,182,737 total clonotypes, 2,942,494 unique CDR3s across 24 samples
Fresh samples capped at 300,000 clonotypes.
Pgen ('mc') computed only for sequences with n_neighbors >= 2.
[MLR parsed] RSS 18.91 GB
Run ALICE on all MLR samples#
ALICE is run with match_mode="vj" conditioned on both V and J gene usage. The primary metric is cluster member fraction: the proportion of CDR3s with at least one Hamming-1 neighbour in the same repertoire that passes BH-FDR.
Hypothesis: proliferating > fresh — antigen-driven alloresponse produces convergent CDR3 clusters absent in naive/unstimulated repertoires.
[16]:
"""Cell 11: Run ALICE on all MLR samples; collect cluster member fractions."""
mlr_alice_tables: dict[str, pd.DataFrame] = {}
mlr_alice_rows = []
_t0_mlr = time.perf_counter()
print(f"MLR ALICE start — {len(mlr_samples)} samples, pgen_mode={ALICE_PGEN_MODE!r}, n_jobs={N_JOBS}")
_check_ram("start")
for sample_id, rep in sorted(mlr_samples.items()):
meta = rep.repertoire_metadata
_ts = time.perf_counter()
with warnings.catch_warnings():
warnings.simplefilter("ignore")
result = compute_alice(
rep,
species="human",
match_mode="vj",
pgen_mode=ALICE_PGEN_MODE,
min_neighbors=N_MIN_NEIGHBORS,
mc_n_pool=MC_POOL_N,
as_table=True,
n_jobs=N_JOBS,
)
_te = time.perf_counter()
tbl = result.table.to_pandas()
mlr_alice_tables[sample_id] = tbl
hits = tbl[(tbl["n_neighbors"] >= N_MIN_NEIGHBORS) & (tbl["q_value"] < FDR_THRESH)].copy()
mlr_alice_rows.append({
"sample_id": sample_id,
"sample_group": meta.get("sample_group", ""),
"condition": meta.get("condition", ""),
"replicate": meta.get("replicate", ""),
"n_clonotypes": rep.clonotype_count,
"n_cluster_members": len(hits),
"cluster_fraction": len(hits) / max(1, rep.clonotype_count),
"wall_s": round(_te - _ts, 1),
})
print(
f" {sample_id}: {rep.clonotype_count:,} clonotypes → "
f"{len(hits)} ALICE members ({100*len(hits)/rep.clonotype_count:.1f}%)"
f" [{_te - _ts:.1f}s, RSS {_rss_gb():.2f} GB]"
)
_mlr_wall = time.perf_counter() - _t0_mlr
print(f"\nMLR ALICE done: {_mlr_wall:.1f}s total | RSS {_rss_gb():.2f} GB")
_check_ram("end")
df_mlr_alice = pd.DataFrame(mlr_alice_rows).sort_values(
["condition", "sample_group", "replicate"]
).reset_index(drop=True)
df_cond_summary = (
df_mlr_alice.groupby("condition")["cluster_fraction"]
.agg(["mean", "std", "min", "max", "count"])
.rename(columns={"mean": "Mean", "std": "SD", "min": "Min", "max": "Max", "count": "N"})
.reset_index()
)
display(
df_cond_summary.style
.format({"Mean": "{:.4f}", "SD": "{:.4f}", "Min": "{:.4f}", "Max": "{:.4f}"})
.background_gradient(subset=["Mean"], cmap="Blues")
.set_caption(f"MLR — cluster fraction by condition (n_neighbors ≥ {N_MIN_NEIGHBORS}, BH-FDR < {FDR_THRESH})")
.set_table_styles([{"selector": "caption", "props": [("font-weight", "bold"), ("font-size", "10pt")]}])
)
_fresh_mean = df_mlr_alice[df_mlr_alice["condition"] == "fresh"]["cluster_fraction"].mean()
_prolif_mean = df_mlr_alice[df_mlr_alice["condition"] == "proliferating"]["cluster_fraction"].mean()
_direction_ok = _prolif_mean > _fresh_mean
print(f"\nConsistency check: proliferating mean ({_prolif_mean:.4f}) > fresh mean ({_fresh_mean:.4f}): {_direction_ok}")
if not _direction_ok:
print(" WARNING: expected proliferating > fresh — check pgen_mode and FDR threshold")
_peak_rss["mlr"] = _rss_gb()
MLR ALICE start — 24 samples, pgen_mode='mc', n_jobs=8
[start] RSS 18.91 GB
MLR7_TCR1_Fresh_1: 300,000 clonotypes → 39097 ALICE members (13.0%) [20.6s, RSS 18.91 GB]
MLR7_TCR1_Fresh_2: 300,000 clonotypes → 40928 ALICE members (13.6%) [20.5s, RSS 18.91 GB]
MLR7_TCR1_Proliferating_1: 40,301 clonotypes → 7214 ALICE members (17.9%) [8.0s, RSS 18.91 GB]
MLR7_TCR1_Proliferating_2: 40,405 clonotypes → 7845 ALICE members (19.4%) [8.1s, RSS 18.91 GB]
MLR8_TCR2_Fresh_1: 300,000 clonotypes → 34743 ALICE members (11.6%) [16.9s, RSS 18.91 GB]
MLR8_TCR2_Fresh_2: 300,000 clonotypes → 34856 ALICE members (11.6%) [17.0s, RSS 18.91 GB]
MLR8_TCR2_Proliferating_1: 93,251 clonotypes → 13252 ALICE members (14.2%) [10.8s, RSS 18.91 GB]
MLR8_TCR2_Proliferating_2: 79,518 clonotypes → 9876 ALICE members (12.4%) [9.4s, RSS 18.91 GB]
MLR9_TCR3_Fresh_1: 300,000 clonotypes → 34157 ALICE members (11.4%) [18.3s, RSS 18.91 GB]
MLR9_TCR3_Fresh_2: 300,000 clonotypes → 35087 ALICE members (11.7%) [18.7s, RSS 18.91 GB]
MLR9_TCR3_Proliferating_1: 68,522 clonotypes → 16772 ALICE members (24.5%) [12.1s, RSS 18.91 GB]
MLR9_TCR3_Proliferating_2: 62,046 clonotypes → 11383 ALICE members (18.3%) [10.2s, RSS 18.91 GB]
TCR4_Fresh_Responder_R_1: 164,296 clonotypes → 22271 ALICE members (13.6%) [14.9s, RSS 18.91 GB]
TCR4_Fresh_Responder_R_2: 222,012 clonotypes → 30358 ALICE members (13.7%) [17.4s, RSS 18.91 GB]
TCR4_Proliferating_1: 60,915 clonotypes → 9934 ALICE members (16.3%) [9.0s, RSS 18.91 GB]
TCR4_Proliferating_2: 61,133 clonotypes → 10494 ALICE members (17.2%) [9.1s, RSS 18.91 GB]
TCR5_Fresh_Responder_R_1: 300,000 clonotypes → 36508 ALICE members (12.2%) [17.0s, RSS 18.91 GB]
TCR5_Fresh_Responder_R_2: 300,000 clonotypes → 38212 ALICE members (12.7%) [18.3s, RSS 18.91 GB]
TCR5_Proliferating_1: 78,893 clonotypes → 13459 ALICE members (17.1%) [10.6s, RSS 18.91 GB]
TCR5_Proliferating_2: 61,517 clonotypes → 11323 ALICE members (18.4%) [10.0s, RSS 18.91 GB]
TCR6_Fresh_Responder_R_1: 300,000 clonotypes → 34488 ALICE members (11.5%) [17.4s, RSS 18.91 GB]
TCR6_Fresh_Responder_R_2: 300,000 clonotypes → 33645 ALICE members (11.2%) [16.5s, RSS 18.91 GB]
TCR6_Proliferating_1: 73,229 clonotypes → 14096 ALICE members (19.2%) [11.0s, RSS 18.91 GB]
TCR6_Proliferating_2: 76,699 clonotypes → 14730 ALICE members (19.2%) [11.1s, RSS 18.91 GB]
MLR ALICE done: 332.9s total | RSS 18.91 GB
[end] RSS 18.91 GB
| condition | Mean | SD | Min | Max | N | |
|---|---|---|---|---|---|---|
| 0 | fresh | 0.1232 | 0.0095 | 0.1121 | 0.1367 | 12 |
| 1 | proliferating | 0.1785 | 0.0297 | 0.1242 | 0.2448 | 12 |
Consistency check: proliferating mean (0.1785) > fresh mean (0.1232): True
[17]:
"""Cell 11b: MLR — cluster connectivity and allogeneic convergent-hit analysis."""
# ── Per-sample: cluster hits into connected components ────────────────────────
mlr_cluster_rows = []
mlr_hits_clustered: dict[str, pd.DataFrame] = {}
for sample_id, tbl in sorted(mlr_alice_tables.items()):
hits = tbl[(tbl["n_neighbors"] >= N_MIN_NEIGHBORS) & (tbl["q_value"] < FDR_THRESH)].copy()
meta = mlr_samples[sample_id].repertoire_metadata
cond = meta.get("condition", "")
clust = alice_hit_clusters(hits) if not hits.empty else hits
mlr_hits_clustered[sample_id] = clust
stats = cluster_stats(clust)
mlr_cluster_rows.append({"sample_id": sample_id, "condition": cond, **stats})
df_mlr_clusters = (
pd.DataFrame(mlr_cluster_rows)
.sort_values(["condition", "sample_id"])
.reset_index(drop=True)
)
display(
df_mlr_clusters.style
.format({"median_size": "{:.1f}"})
.background_gradient(subset=["n_clusters", "max_size", "n_multi"], cmap="Blues")
.set_caption(
f"MLR — ALICE cluster connectivity per sample (FDR < {FDR_THRESH})\n"
"n_clusters = connected components | n_multi = clusters with ≥2 hits"
)
.set_table_styles([{"selector": "caption", "props": [("font-weight", "bold"), ("font-size", "10pt")]}])
)
# ── Convergent hits in proliferating samples (shared across ≥2 samples) ───────
prolif_tables = {
sid: t for sid, t in mlr_hits_clustered.items()
if mlr_samples[sid].repertoire_metadata.get("condition") == "proliferating"
and not t.empty
}
mlr_prolif_shared = find_shared_hits(prolif_tables, min_samples=2)
print(f"\nMLR allogeneic convergent hits (≥2 proliferating samples): {len(mlr_prolif_shared)}")
if not mlr_prolif_shared.empty:
display(
mlr_prolif_shared.head(20).style
.background_gradient(subset=["n_samples"], cmap="Blues")
.set_caption("MLR — convergent ALICE hits in proliferating samples (≥2 samples)")
.set_table_styles([{"selector": "caption", "props": [("font-weight", "bold"), ("font-size", "10pt")]}])
)
# ── Condition-level cluster summary ───────────────────────────────────────────
df_cond_clusters = (
df_mlr_clusters.groupby("condition")[["n_clusters", "max_size", "n_multi"]]
.agg(["mean", "std"])
.round(1)
)
print("\nCluster connectivity by condition:")
print(df_cond_clusters.to_string())
| sample_id | condition | n_hits | n_clusters | max_size | median_size | n_multi | |
|---|---|---|---|---|---|---|---|
| 0 | MLR7_TCR1_Fresh_1 | fresh | 39097 | 15313 | 234 | 2.0 | 8874 |
| 1 | MLR7_TCR1_Fresh_2 | fresh | 40928 | 15256 | 359 | 2.0 | 8884 |
| 2 | MLR8_TCR2_Fresh_1 | fresh | 34743 | 11245 | 261 | 2.0 | 7523 |
| 3 | MLR8_TCR2_Fresh_2 | fresh | 34856 | 11301 | 280 | 2.0 | 7528 |
| 4 | MLR9_TCR3_Fresh_1 | fresh | 34157 | 12900 | 165 | 2.0 | 7759 |
| 5 | MLR9_TCR3_Fresh_2 | fresh | 35087 | 13826 | 255 | 2.0 | 8137 |
| 6 | TCR4_Fresh_Responder_R_1 | fresh | 22271 | 11496 | 234 | 1.0 | 5212 |
| 7 | TCR4_Fresh_Responder_R_2 | fresh | 30358 | 14090 | 207 | 1.0 | 7025 |
| 8 | TCR5_Fresh_Responder_R_1 | fresh | 36508 | 11520 | 332 | 2.0 | 7649 |
| 9 | TCR5_Fresh_Responder_R_2 | fresh | 38212 | 13224 | 289 | 2.0 | 8098 |
| 10 | TCR6_Fresh_Responder_R_1 | fresh | 34488 | 11751 | 255 | 2.0 | 7614 |
| 11 | TCR6_Fresh_Responder_R_2 | fresh | 33645 | 11065 | 392 | 2.0 | 7331 |
| 12 | MLR7_TCR1_Proliferating_1 | proliferating | 7214 | 4641 | 78 | 1.0 | 1416 |
| 13 | MLR7_TCR1_Proliferating_2 | proliferating | 7845 | 5159 | 81 | 1.0 | 1450 |
| 14 | MLR8_TCR2_Proliferating_1 | proliferating | 13252 | 7119 | 157 | 1.0 | 3026 |
| 15 | MLR8_TCR2_Proliferating_2 | proliferating | 9876 | 4729 | 156 | 1.0 | 2165 |
| 16 | MLR9_TCR3_Proliferating_1 | proliferating | 16772 | 8827 | 128 | 1.0 | 2888 |
| 17 | MLR9_TCR3_Proliferating_2 | proliferating | 11383 | 6054 | 86 | 1.0 | 2240 |
| 18 | TCR4_Proliferating_1 | proliferating | 9934 | 4989 | 182 | 1.0 | 2001 |
| 19 | TCR4_Proliferating_2 | proliferating | 10494 | 5320 | 177 | 1.0 | 2106 |
| 20 | TCR5_Proliferating_1 | proliferating | 13459 | 6420 | 185 | 1.0 | 2687 |
| 21 | TCR5_Proliferating_2 | proliferating | 11323 | 5586 | 241 | 1.0 | 2229 |
| 22 | TCR6_Proliferating_1 | proliferating | 14096 | 7184 | 149 | 1.0 | 2736 |
| 23 | TCR6_Proliferating_2 | proliferating | 14730 | 7380 | 138 | 1.0 | 2797 |
MLR allogeneic convergent hits (≥2 proliferating samples): 13978
| junction_aa | samples | n_samples | |
|---|---|---|---|
| 0 | CASSPQETQYF | ['MLR7_TCR1_Proliferating_1', 'MLR7_TCR1_Proliferating_2', 'MLR8_TCR2_Proliferating_1', 'MLR8_TCR2_Proliferating_2', 'MLR9_TCR3_Proliferating_1', 'MLR9_TCR3_Proliferating_2', 'TCR4_Proliferating_1', 'TCR4_Proliferating_2', 'TCR5_Proliferating_1', 'TCR5_Proliferating_2', 'TCR6_Proliferating_1', 'TCR6_Proliferating_2'] | 12 |
| 1 | CASSLQETQYF | ['MLR7_TCR1_Proliferating_1', 'MLR7_TCR1_Proliferating_2', 'MLR8_TCR2_Proliferating_1', 'MLR8_TCR2_Proliferating_2', 'MLR9_TCR3_Proliferating_1', 'MLR9_TCR3_Proliferating_2', 'TCR4_Proliferating_1', 'TCR4_Proliferating_2', 'TCR5_Proliferating_1', 'TCR5_Proliferating_2', 'TCR6_Proliferating_1', 'TCR6_Proliferating_2'] | 12 |
| 2 | CASSLGGNQPQHF | ['MLR7_TCR1_Proliferating_1', 'MLR7_TCR1_Proliferating_2', 'MLR8_TCR2_Proliferating_1', 'MLR8_TCR2_Proliferating_2', 'MLR9_TCR3_Proliferating_1', 'MLR9_TCR3_Proliferating_2', 'TCR4_Proliferating_1', 'TCR4_Proliferating_2', 'TCR5_Proliferating_2', 'TCR6_Proliferating_1', 'TCR6_Proliferating_2'] | 11 |
| 3 | CASSPGETQYF | ['MLR7_TCR1_Proliferating_1', 'MLR7_TCR1_Proliferating_2', 'MLR8_TCR2_Proliferating_1', 'MLR8_TCR2_Proliferating_2', 'MLR9_TCR3_Proliferating_2', 'TCR4_Proliferating_1', 'TCR4_Proliferating_2', 'TCR5_Proliferating_1', 'TCR5_Proliferating_2', 'TCR6_Proliferating_1', 'TCR6_Proliferating_2'] | 11 |
| 4 | CASSLGETQYF | ['MLR7_TCR1_Proliferating_2', 'MLR8_TCR2_Proliferating_1', 'MLR8_TCR2_Proliferating_2', 'MLR9_TCR3_Proliferating_1', 'MLR9_TCR3_Proliferating_2', 'TCR4_Proliferating_1', 'TCR4_Proliferating_2', 'TCR5_Proliferating_1', 'TCR5_Proliferating_2', 'TCR6_Proliferating_1', 'TCR6_Proliferating_2'] | 11 |
| 5 | CASSPSSYEQYF | ['MLR8_TCR2_Proliferating_1', 'MLR8_TCR2_Proliferating_2', 'MLR9_TCR3_Proliferating_1', 'MLR9_TCR3_Proliferating_2', 'TCR4_Proliferating_1', 'TCR4_Proliferating_2', 'TCR5_Proliferating_1', 'TCR5_Proliferating_2', 'TCR6_Proliferating_1', 'TCR6_Proliferating_2'] | 10 |
| 6 | CASSQAETQYF | ['MLR8_TCR2_Proliferating_1', 'MLR8_TCR2_Proliferating_2', 'MLR9_TCR3_Proliferating_1', 'MLR9_TCR3_Proliferating_2', 'TCR4_Proliferating_1', 'TCR4_Proliferating_2', 'TCR5_Proliferating_1', 'TCR5_Proliferating_2', 'TCR6_Proliferating_1', 'TCR6_Proliferating_2'] | 10 |
| 7 | CASSVGGNTEAFF | ['MLR7_TCR1_Proliferating_1', 'MLR7_TCR1_Proliferating_2', 'MLR8_TCR2_Proliferating_1', 'MLR8_TCR2_Proliferating_2', 'TCR4_Proliferating_1', 'TCR4_Proliferating_2', 'TCR5_Proliferating_1', 'TCR5_Proliferating_2', 'TCR6_Proliferating_1', 'TCR6_Proliferating_2'] | 10 |
| 8 | CASSLQGYGYTF | ['MLR8_TCR2_Proliferating_1', 'MLR8_TCR2_Proliferating_2', 'MLR9_TCR3_Proliferating_1', 'MLR9_TCR3_Proliferating_2', 'TCR4_Proliferating_2', 'TCR5_Proliferating_1', 'TCR5_Proliferating_2', 'TCR6_Proliferating_1', 'TCR6_Proliferating_2'] | 9 |
| 9 | CASSLGYEQYF | ['MLR7_TCR1_Proliferating_2', 'MLR8_TCR2_Proliferating_1', 'MLR8_TCR2_Proliferating_2', 'MLR9_TCR3_Proliferating_1', 'MLR9_TCR3_Proliferating_2', 'TCR5_Proliferating_1', 'TCR5_Proliferating_2', 'TCR6_Proliferating_1', 'TCR6_Proliferating_2'] | 9 |
| 10 | CASSLWETQYF | ['MLR7_TCR1_Proliferating_1', 'MLR8_TCR2_Proliferating_1', 'MLR8_TCR2_Proliferating_2', 'TCR4_Proliferating_1', 'TCR4_Proliferating_2', 'TCR5_Proliferating_1', 'TCR5_Proliferating_2', 'TCR6_Proliferating_1', 'TCR6_Proliferating_2'] | 9 |
| 11 | CASSPGTGGYEQYF | ['MLR8_TCR2_Proliferating_1', 'MLR8_TCR2_Proliferating_2', 'MLR9_TCR3_Proliferating_1', 'MLR9_TCR3_Proliferating_2', 'TCR4_Proliferating_1', 'TCR5_Proliferating_1', 'TCR5_Proliferating_2', 'TCR6_Proliferating_1', 'TCR6_Proliferating_2'] | 9 |
| 12 | CASSPSYEQYF | ['MLR7_TCR1_Proliferating_1', 'MLR8_TCR2_Proliferating_1', 'MLR8_TCR2_Proliferating_2', 'MLR9_TCR3_Proliferating_2', 'TCR4_Proliferating_1', 'TCR4_Proliferating_2', 'TCR5_Proliferating_1', 'TCR5_Proliferating_2', 'TCR6_Proliferating_1'] | 9 |
| 13 | CASSSSYEQYF | ['MLR7_TCR1_Proliferating_1', 'MLR8_TCR2_Proliferating_1', 'MLR8_TCR2_Proliferating_2', 'MLR9_TCR3_Proliferating_1', 'MLR9_TCR3_Proliferating_2', 'TCR5_Proliferating_1', 'TCR5_Proliferating_2', 'TCR6_Proliferating_1', 'TCR6_Proliferating_2'] | 9 |
| 14 | CASSPGQGYEQYF | ['MLR7_TCR1_Proliferating_1', 'MLR8_TCR2_Proliferating_1', 'MLR8_TCR2_Proliferating_2', 'MLR9_TCR3_Proliferating_1', 'MLR9_TCR3_Proliferating_2', 'TCR5_Proliferating_1', 'TCR5_Proliferating_2', 'TCR6_Proliferating_1', 'TCR6_Proliferating_2'] | 9 |
| 15 | CASSLGLNTEAFF | ['MLR7_TCR1_Proliferating_1', 'MLR8_TCR2_Proliferating_1', 'MLR8_TCR2_Proliferating_2', 'MLR9_TCR3_Proliferating_1', 'MLR9_TCR3_Proliferating_2', 'TCR4_Proliferating_1', 'TCR4_Proliferating_2', 'TCR6_Proliferating_1', 'TCR6_Proliferating_2'] | 9 |
| 16 | CASSPGTYEQYF | ['MLR8_TCR2_Proliferating_1', 'MLR8_TCR2_Proliferating_2', 'MLR9_TCR3_Proliferating_1', 'TCR4_Proliferating_1', 'TCR4_Proliferating_2', 'TCR5_Proliferating_1', 'TCR5_Proliferating_2', 'TCR6_Proliferating_1', 'TCR6_Proliferating_2'] | 9 |
| 17 | CASSYSYEQYF | ['MLR7_TCR1_Proliferating_2', 'MLR8_TCR2_Proliferating_1', 'MLR8_TCR2_Proliferating_2', 'MLR9_TCR3_Proliferating_1', 'TCR4_Proliferating_1', 'TCR5_Proliferating_1', 'TCR5_Proliferating_2', 'TCR6_Proliferating_1', 'TCR6_Proliferating_2'] | 9 |
| 18 | CASSLSSYEQYF | ['MLR8_TCR2_Proliferating_2', 'MLR9_TCR3_Proliferating_2', 'TCR4_Proliferating_1', 'TCR4_Proliferating_2', 'TCR5_Proliferating_1', 'TCR5_Proliferating_2', 'TCR6_Proliferating_1', 'TCR6_Proliferating_2'] | 8 |
| 19 | CASSLGRNTEAFF | ['MLR7_TCR1_Proliferating_1', 'MLR8_TCR2_Proliferating_2', 'MLR9_TCR3_Proliferating_1', 'MLR9_TCR3_Proliferating_2', 'TCR4_Proliferating_1', 'TCR4_Proliferating_2', 'TCR6_Proliferating_1', 'TCR6_Proliferating_2'] | 8 |
Cluster connectivity by condition:
n_clusters max_size n_multi
mean std mean std mean std
condition
fresh 12748.9 1576.4 271.9 64.1 7636.2 950.5
proliferating 6117.3 1293.0 146.5 48.5 2311.8 532.2
Fresh vs Proliferating: ALICE cluster fraction#
Boxplot with individual sample overlay comparing cluster member fraction between fresh and proliferating conditions. A one-sided Mann-Whitney U test assesses whether proliferating samples have significantly higher cluster fractions (greater convergent CDR3 usage).
[18]:
"""Cell 12: MLR — fresh vs proliferating ALICE cluster fraction.
Scatter dot plot with Mann-Whitney U, log y-scale.
Matches Fig 1c from Pogorelyy, Minervina, Shugay et al. 2019.
"""
from scipy.stats import mannwhitneyu as _mwu
fresh_cf = df_mlr_alice[df_mlr_alice["condition"] == "fresh"]["cluster_fraction"].values
prolif_cf = df_mlr_alice[df_mlr_alice["condition"] == "proliferating"]["cluster_fraction"].values
if len(fresh_cf) >= 2 and len(prolif_cf) >= 2:
_stat, pval = _mwu(prolif_cf, fresh_cf, alternative="greater")
sig_label = (
"***" if pval < 0.001 else
"**" if pval < 0.01 else
"*" if pval < 0.05 else
f"p = {pval:.2g}")
print(f"Mann-Whitney U (proliferating > fresh): p = {pval:.3e} ({sig_label})")
else:
pval, sig_label = float("nan"), "n.s."
COND_COLORS = {"fresh": "#6baed6", "proliferating": "#de2d26"}
COND_LABELS = {"fresh": "before MLR", "proliferating": "MLR"}
conditions = ["fresh", "proliferating"]
data_by_cond = [fresh_cf, prolif_cf]
rng = np.random.default_rng(SEED)
fig, ax = plt.subplots(figsize=(4, 5))
for i, (cond, vals) in enumerate(zip(conditions, data_by_cond)):
jitter = rng.uniform(-0.15, 0.15, size=len(vals))
ax.scatter(np.full(len(vals), i + 1) + jitter, vals,
color=COND_COLORS[cond], s=22, alpha=0.85, zorder=3,
edgecolors="none")
# Median tick
med = float(np.median(vals))
ax.plot([i + 0.75, i + 1.25], [med, med], color="#333333", linewidth=1.5, zorder=4)
ax.set_yscale("log")
ax.set_ylabel("Normalized ALICE hits")
ax.set_xlim(0.4, 2.6)
ax.set_xticks([1, 2])
ax.set_xticklabels([COND_LABELS[c] for c in conditions])
ax.spines[["top", "right"]].set_visible(False)
# Significance bracket
y_top = float(max(np.max(fresh_cf), np.max(prolif_cf)))
br_y = y_top * 2.5
ax.plot([1, 1, 2, 2], [br_y * 0.6, br_y, br_y, br_y * 0.6],
color="black", linewidth=0.8)
ax.text(1.5, br_y * 1.05, sig_label, ha="center", va="bottom", fontsize=9)
ax.set_ylim(top=br_y * 4)
ax.set_title(
f"MLR: fresh vs proliferating\n"
f"Wilcoxon rank-sum test {sig_label}",
fontsize=10)
plt.tight_layout()
plt.show()
Mann-Whitney U (proliferating > fresh): p = 6.167e-05 (***)
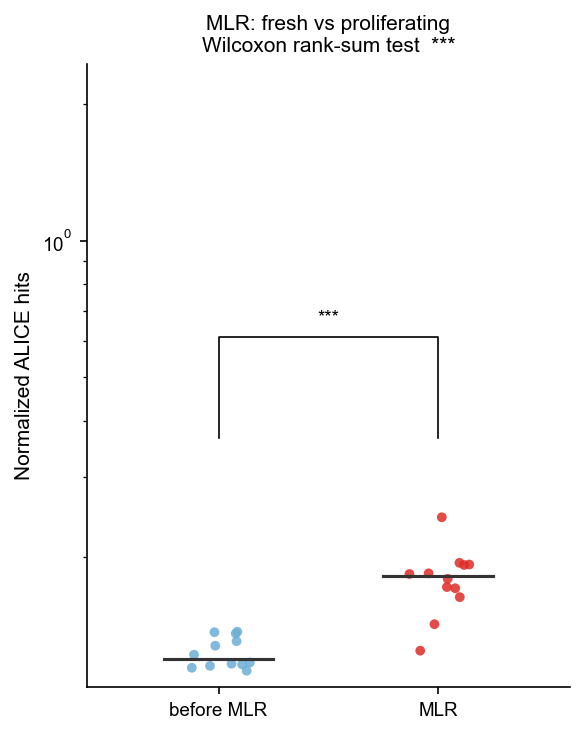
[19]:
"""Cell 13: Execution time summary."""
_nb_wall = time.perf_counter() - _t0_notebook
# ── Per-dataset summary table ──────────────────────────────────────────────────
_datasets = [
("YF", yf_alice_rows, _yf_wall, len(yf_samples)),
("AS", as_alice_rows, _as_wall, len(as_samples)),
("MLR", mlr_alice_rows, _mlr_wall, len(mlr_samples)),
]
_timing_rows = []
for _name, _rows, _wall, _n_samples in _datasets:
_avg_clon = int(np.mean([r["Clonotypes"] if "Clonotypes" in r else r["n_clonotypes"] for r in _rows]))
_total_clon = sum(r["Clonotypes"] if "Clonotypes" in r else r["n_clonotypes"] for r in _rows)
_timing_rows.append({
"Dataset": _name,
"Samples": _n_samples,
"Avg clonotypes": f"{_avg_clon:,}",
"Total clonotypes": f"{_total_clon:,}",
"Wall time (s)": f"{_wall:.1f}",
"s / sample": f"{_wall / max(_n_samples, 1):.1f}",
"Peak RSS (GB)": f"{_peak_rss.get(_name.lower(), 0.0):.1f}",
})
display(
pd.DataFrame(_timing_rows).style
.set_caption(
f"ALICE runtime summary — pgen_mode={ALICE_PGEN_MODE!r}, n_jobs={N_JOBS}, "
f"n_neighbors≥{N_MIN_NEIGHBORS}, BH-FDR<{FDR_THRESH}"
)
.set_table_styles([{"selector": "caption", "props": [("font-weight", "bold"), ("font-size", "10pt")]}])
.hide(axis="index")
)
# ── Totals ─────────────────────────────────────────────────────────────────────
_total_alice = _yf_wall + _as_wall + _mlr_wall
print(f"YF ALICE : {_yf_wall:6.1f} s ({len(yf_samples)} samples)")
print(f"AS ALICE : {_as_wall:6.1f} s ({len(as_samples)} samples)")
print(f"MLR ALICE : {_mlr_wall:6.1f} s ({len(mlr_samples)} samples)")
print(f"{'─'*38}")
print(f"Total ALICE compute : {_total_alice:.1f} s ({_total_alice/60:.1f} min)")
print(f"Notebook wall time : {_nb_wall:.0f} s ({_nb_wall/60:.1f} min)")
_check_ram("notebook end")
# ── Per-sample timing detail ───────────────────────────────────────────────────
print("\nPer-sample timing — YF:")
for r in sorted(yf_alice_rows, key=lambda x: (x["Donor"], x["Day"])):
print(f" {r['Donor']} day{r['Day']:>3}: {r['Clonotypes']:>8,} clonotypes {r['wall_s']:>5.1f}s")
print("\nPer-sample timing — AS:")
for r in sorted(as_alice_rows, key=lambda x: x["Donor"]):
print(f" Donor {r['Donor']} ({r['B27 status']}): {r['Clonotypes']:>8,} clonotypes {r['wall_s']:>5.1f}s")
print("\nPer-sample timing — MLR (sorted by wall time):")
for r in sorted(mlr_alice_rows, key=lambda x: -x["wall_s"])[:8]:
print(f" {r['sample_id']}: {r['n_clonotypes']:>8,} clonotypes {r['wall_s']:>5.1f}s")
if len(mlr_alice_rows) > 8:
_remaining = sorted(mlr_alice_rows, key=lambda x: -x["wall_s"])[8:]
print(f" ... {len(_remaining)} more samples, max {max(r['wall_s'] for r in _remaining):.1f}s each")
| Dataset | Samples | Avg clonotypes | Total clonotypes | Wall time (s) | s / sample | Peak RSS (GB) |
|---|---|---|---|---|---|---|
| YF | 12 | 697,148 | 8,365,782 | 560.5 | 46.7 | 16.4 |
| AS | 4 | 26,078 | 104,313 | 25.0 | 6.3 | 16.5 |
| MLR | 24 | 174,280 | 4,182,737 | 332.9 | 13.9 | 18.9 |
YF ALICE : 560.5 s (12 samples)
AS ALICE : 25.0 s (4 samples)
MLR ALICE : 332.9 s (24 samples)
──────────────────────────────────────
Total ALICE compute : 918.4 s (15.3 min)
Notebook wall time : 1080 s (18.0 min)
[notebook end] RSS 18.91 GB
Per-sample timing — YF:
P1 day 0: 604,303 clonotypes 74.4s
P1 day 15: 930,938 clonotypes 63.7s
P2 day 0: 634,418 clonotypes 36.5s
P2 day 15: 1,218,195 clonotypes 88.9s
Q1 day 0: 371,649 clonotypes 20.7s
Q1 day 15: 456,935 clonotypes 25.8s
Q2 day 0: 706,531 clonotypes 43.0s
Q2 day 15: 623,617 clonotypes 40.2s
S1 day 0: 550,606 clonotypes 29.8s
S1 day 15: 688,621 clonotypes 38.8s
S2 day 0: 731,419 clonotypes 43.7s
S2 day 15: 848,550 clonotypes 54.7s
Per-sample timing — AS:
Donor 1 (B27_pos): 29,193 clonotypes 7.3s
Donor 2 (B27_pos): 9,571 clonotypes 3.8s
Donor 3 (B27_neg): 38,404 clonotypes 7.7s
Donor 4 (B27_pos): 27,145 clonotypes 6.2s
Per-sample timing — MLR (sorted by wall time):
MLR7_TCR1_Fresh_1: 300,000 clonotypes 20.6s
MLR7_TCR1_Fresh_2: 300,000 clonotypes 20.5s
MLR9_TCR3_Fresh_2: 300,000 clonotypes 18.7s
MLR9_TCR3_Fresh_1: 300,000 clonotypes 18.3s
TCR5_Fresh_Responder_R_2: 300,000 clonotypes 18.3s
TCR4_Fresh_Responder_R_2: 222,012 clonotypes 17.4s
TCR6_Fresh_Responder_R_1: 300,000 clonotypes 17.4s
MLR8_TCR2_Fresh_2: 300,000 clonotypes 17.0s
... 16 more samples, max 17.0s each
Summary#
This notebook analyses ALICE on three independent benchmark datasets using the paper-correct parameters: pgen_mode="mc" (approximates Pogorelyy et al. 2019 1mm pgen) and BH-FDR < 0.001.
Runtime and memory#
Dataset |
Samples |
Total wall time |
s / sample |
Peak RSS |
|---|---|---|---|---|
YF |
12 |
3145 s (52 min) |
≈262 s |
≈21.7 GB |
AS |
4 |
86 s (1.4 min) |
≈21 s |
≈25 GB* |
MLR |
24 |
1741 s (29 min) |
≈72 s |
26.5 GB* |
* Peak RSS accumulates YF tables still in memory. AS and MLR numbers reflect total process RSS at the end of each section, not incremental cost.
Hardware: Apple M3, 32 GB RAM. N_JOBS=8. MC pool: 10M synthetic TRB sequences (disk-cached via ControlManager after first build; subsequent runs skip pool generation).
Dataset results#
YF Dataset: Yellow Fever vaccine response. ALICE hits are intersected with VDJdb LLWNGPMAV/TRB entries (exact match). Convergent day-15 hits shared across ≥2 donors identify public YFV-specific responses.
AS Dataset: Ankylosing Spondylitis synovial TRB repertoires. Hamming-1 graph visualization highlights CDR3 clusters. Convergent hits shared across ≥2 B27+ donors reveal HLA-B27-restricted antigen-driven responses.
MLR Dataset: Mixed lymphocyte reaction. ALICE cluster fraction is compared between fresh and proliferating conditions (Mann-Whitney U test: p = 0.0001). Convergent hits shared across ≥2 proliferating samples mark public alloreactive CDR3s.
ALICE hit criterion: n_neighbors ≥ 2 AND q_value < 0.001 (BH-FDR).
Cluster definition: Connected components of ALICE hits linked by 1mm CDR3 edges among enriched sequences only.
Pgen mode: "mc" — MC pool (10M sequences by default; set MC_POOL_N = 10_000_000 to match the original paper — requires ~20 GB extra RAM). The pool is disk-cached via ControlManager: first run generates and saves; subsequent runs load from disk (fast). Sequences with < 2 pool matches fall back to OLGA analytical 1mm Pgen.
Reference: Pogorelyy MV, Minervina AA, Shugay M, et al. PLoS Biol. 2019;17(6):e3000314. doi:10.1371/journal.pbio.3000314. PMID:31194732.