CDR3 Graph Analysis#
[1]:
import sys
import time
from pathlib import Path
import igraph as ig
import matplotlib.pyplot as plt
import pandas as pd
repo_root = Path.cwd().resolve().parent if Path.cwd().name == "notebooks" else Path.cwd().resolve()
if str(repo_root) not in sys.path:
sys.path.insert(0, str(repo_root))
from mir.common.clonotype import Clonotype
from mir.graph.edit_distance_graph import build_edit_distance_graph
from mir.utils.notebook_assets import ensure_airr_benchmark, find_airr_benchmark_vdjdb_slim
/Users/mikesh/vcs/mirpy/.venv/lib/python3.12/site-packages/tqdm/auto.py:21: TqdmWarning: IProgress not found. Please update jupyter and ipywidgets. See https://ipywidgets.readthedocs.io/en/stable/user_install.html
from .autonotebook import tqdm as notebook_tqdm
[2]:
benchmark_root = ensure_airr_benchmark(repo_root, allow_patterns=["vdjdb/**"])
vdjdb_path = find_airr_benchmark_vdjdb_slim(benchmark_root)
vdjdb = pd.read_csv(vdjdb_path, sep='\t')
vdjdb = vdjdb[vdjdb['gene'].eq('TRB')].copy()
print(f'Loaded VDJdb snapshot from: {vdjdb_path}')
print(vdjdb.head())
vdjdb['antigen.epitope'].value_counts().head(20)
Loaded VDJdb snapshot from: /Users/mikesh/vcs/mirpy/notebooks/assets/large/airr_benchmark/vdjdb/vdjdb-2025-12-29/vdjdb.slim.txt.gz
gene cdr3 species antigen.epitope antigen.gene \
50093 TRB ATSIRFTDTQYF HomoSapiens PQPELPYPQPQ Gliadin
50094 TRB CAAADEEIGNQPQHF HomoSapiens ATDALMTGY NS3
50095 TRB CAAAERNTGELFF HomoSapiens YLQPRTFLL Spike
50096 TRB CAAAGTASTDTQYF HomoSapiens RAKFKQLL BZLF1
50097 TRB CAAAQGYEQYF HomoSapiens NLVPMVATV pp65
antigen.species complex.id v.segm j.segm mhc.a \
50093 TriticumAestivum 0 TRBV7-2*01 TRBJ2-3*01 HLA-DQA1*05:01
50094 HCV 0 TRBV10-3*01 TRBJ1-5*01 HLA-A*01:01
50095 SARS-CoV-2 0 TRBV28*01 TRBJ2-2*01 HLA-A*02
50096 EBV 80505,80553 TRBV10-3*01 TRBJ2-3*01 HLA-B*08:01
50097 CMV 71852 TRBV2*01 TRBJ2-7*01 HLA-A*02:01
mhc.b mhc.class \
50093 HLA-DQB1*02:01 MHCII
50094 B2M MHCI
50095 B2M MHCI
50096 B2M MHCI
50097 B2M MHCI
reference.id vdjdb.score \
50093 PMID:23878218 0
50094 PMID:21160049 1
50095 https://github.com/antigenomics/vdjdb-db/issue... 0
50096 PMID:34811538 0
50097 PMID:39746936 0
vdjdb.pgen.score TCR_hash \
50093 0 NaN
50094 1 NaN
50095 2 NaN
50096 2 061647eb1cdc13de5aa6a5192b950ba6cd1233ffa6c871...
50097 2 c3160eeb230051a6139814a7038406ee0a79d86cde0b0d...
j.start v.end
50093 6 -1
50094 9 2
50095 6 2
50096 7 2
50097 6 2
[2]:
antigen.epitope
SLLMWITQV 29698
NLVPMVATV 13044
KLGGALQAK 12667
GILGFVFTL 5236
VEALYLVCG 2545
AVFDRKSDAK 1648
ELAGIGILTV 1431
RAKFKQLL 1410
GLCTLVAML 1227
YLQPRTFLL 903
LLAGIGTVPI 885
VEALYLVSG 853
IVTDFSVIK 551
SPRWYFYYL 545
CTPYDINQM 530
TTPESANL 523
KRWIILGLNK 469
TFEYVSQPFLMDLE 445
TTDPSFLGRY 438
YVLDHLIVV 435
Name: count, dtype: int64
[3]:
def get_seqs(epitopes: list[str], size: int = 500, gene: str = 'TRB') -> pd.DataFrame:
rows = vdjdb[vdjdb['gene'].eq(gene) & vdjdb['antigen.epitope'].isin(epitopes)].copy()
sampled_parts = []
for epitope, grp in rows.groupby('antigen.epitope'):
n_take = min(size, len(grp))
sampled = grp.sample(n=n_take, random_state=42).copy()
sampled['antigen.epitope'] = epitope
sampled_parts.append(sampled)
if not sampled_parts:
return pd.DataFrame(columns=['junction_aa', 'antigen.epitope'])
sampled = pd.concat(sampled_parts, ignore_index=True).drop_duplicates(subset=['cdr3'])
return sampled[['cdr3', 'antigen.epitope']].rename(columns={'cdr3': 'junction_aa'})
epitopes = ['GLCTLVAML', 'GILGFVFTL', 'NLVPMVATV']
df_epi = get_seqs(epitopes=epitopes, size=1000)
print(df_epi['antigen.epitope'].value_counts())
print('Unique CDR3:', df_epi['junction_aa'].nunique())
antigen.epitope
GILGFVFTL 1000
NLVPMVATV 999
GLCTLVAML 991
Name: count, dtype: int64
Unique CDR3: 2990
[4]:
clonotypes = [
Clonotype(
sequence_id=str(i),
locus='TRB',
junction_aa=aa,
v_gene='TRBV7-9*01',
j_gene='TRBJ2-1*01',
duplicate_count=1,
_validate=False,
)
for i, aa in enumerate(df_epi['junction_aa'].tolist())
]
start = time.perf_counter()
g_serial = build_edit_distance_graph(clonotypes, metric='hamming', threshold=1, n_jobs=1)
t_serial = time.perf_counter() - start
print('Serial hamming runtime:', round(t_serial, 3), 's')
start = time.perf_counter()
g_parallel = build_edit_distance_graph(clonotypes, metric='hamming', threshold=1, n_jobs=4)
t_parallel = time.perf_counter() - start
print('Parallel (n_jobs=4) hamming runtime:', round(t_parallel, 3), 's')
if t_parallel > 0:
print('Speedup:', round(t_serial / t_parallel, 2), 'x')
assert g_serial.ecount() == g_parallel.ecount()
graph = g_parallel
print('Vertices:', graph.vcount(), 'Edges:', graph.ecount())
Serial hamming runtime: 0.023 s
Parallel (n_jobs=4) hamming runtime: 2.452 s
Speedup: 0.01 x
Vertices: 2990 Edges: 623
[5]:
start = time.perf_counter()
layout = graph.layout_graphopt(niter=2000, node_charge=0.005)
elapsed = time.perf_counter() - start
components = graph.components()
print('Layout runtime:', round(elapsed, 3), 's')
print('Connected components:', len(components))
print('Largest CC size:', max(components.sizes()) if components.sizes() else 0)
Layout runtime: 15.337 s
Connected components: 2567
Largest CC size: 65
[6]:
coords = pd.DataFrame(layout.coords, columns=['x', 'y'])
df_graph = pd.DataFrame({
'sequence_id': [str(i) for i in range(graph.vcount())],
'junction_aa': graph.vs['name'],
'component_id': graph.components().membership,
'degree': graph.degree(),
})
df_graph = pd.concat([df_graph, coords], axis=1)
print(df_graph.head())
sequence_id junction_aa component_id degree x \
0 0 CASSPRQGWSQETQYF 0 0 2402.128081
1 1 CASSIFTIEPQHF 1 0 1692.071181
2 2 CASRSLAGVWDTQYF 2 0 -1539.777416
3 3 CASSLTVAGVYPGYEQYF 3 0 -2136.915199
4 4 CASSNRAAYEQYF 4 2 349.640074
y
0 -2246.509161
1 -2311.982019
2 -3258.958664
3 -1248.494955
4 -121.086847
[7]:
df_graph_summary = (
df_graph.groupby('component_id', as_index=False)
.agg(
cluster_size=('component_id', 'size'),
x_mean=('x', 'mean'),
y_mean=('y', 'mean'),
mean_degree=('degree', 'mean'),
)
)
print(df_graph_summary.head(10))
component_id cluster_size x_mean y_mean mean_degree
0 0 1 2402.128081 -2246.509161 0.000000
1 1 1 1692.071181 -2311.982019 0.000000
2 2 1 -1539.777416 -3258.958664 0.000000
3 3 1 -2136.915199 -1248.494955 0.000000
4 4 65 -65.944975 187.387491 3.692308
5 5 1 -1357.619093 3467.835778 0.000000
6 6 1 -2324.450880 331.797380 0.000000
7 7 1 1302.895952 -2213.111617 0.000000
8 8 2 -83.579680 -1525.636353 1.000000
9 9 1 -510.599900 2254.446718 0.000000
[8]:
df_annot = df_epi.drop_duplicates(subset=['junction_aa'])
df_plot = (
df_graph.merge(df_graph_summary, on='component_id', how='left')
.merge(df_annot, on='junction_aa', how='left')
)
print(df_plot.head())
sequence_id junction_aa component_id degree x \
0 0 CASSPRQGWSQETQYF 0 0 2402.128081
1 1 CASSIFTIEPQHF 1 0 1692.071181
2 2 CASRSLAGVWDTQYF 2 0 -1539.777416
3 3 CASSLTVAGVYPGYEQYF 3 0 -2136.915199
4 4 CASSNRAAYEQYF 4 2 349.640074
y cluster_size x_mean y_mean mean_degree \
0 -2246.509161 1 2402.128081 -2246.509161 0.000000
1 -2311.982019 1 1692.071181 -2311.982019 0.000000
2 -3258.958664 1 -1539.777416 -3258.958664 0.000000
3 -1248.494955 1 -2136.915199 -1248.494955 0.000000
4 -121.086847 65 -65.944975 187.387491 3.692308
antigen.epitope
0 GILGFVFTL
1 GILGFVFTL
2 GILGFVFTL
3 GILGFVFTL
4 GILGFVFTL
[9]:
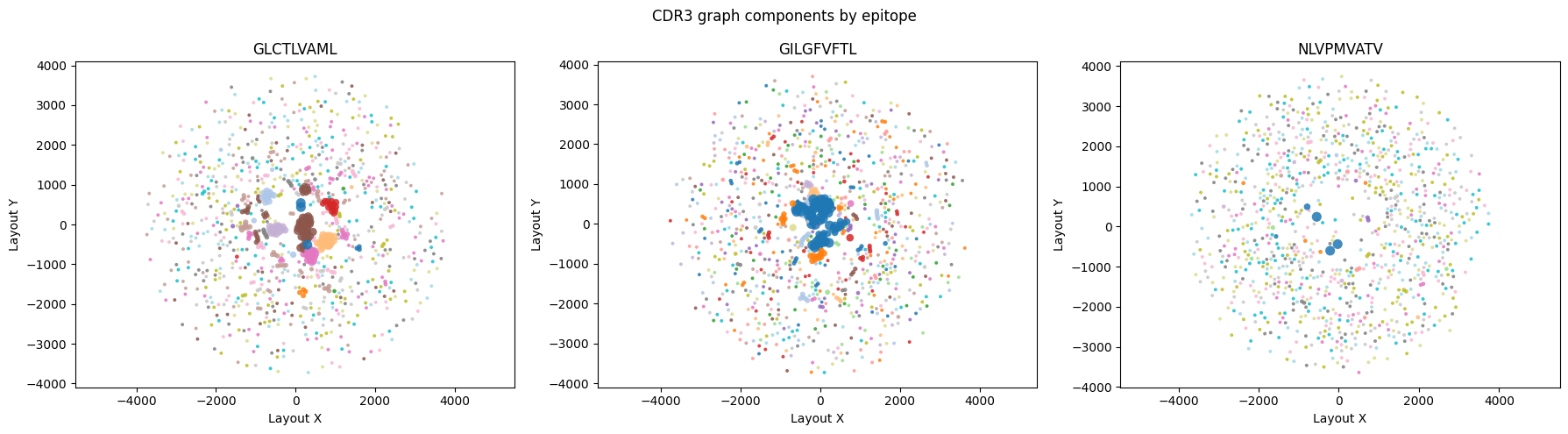
fig, axes = plt.subplots(1, len(epitopes), figsize=(6 * len(epitopes), 5), squeeze=False)
for ax, epitope in zip(axes[0], epitopes):
sub = df_plot[df_plot['antigen.epitope'].eq(epitope)].copy()
if sub.empty:
ax.set_title(f'{epitope} (no data)')
ax.axis('off')
continue
sizes = (sub['cluster_size'].clip(lower=1) ** 0.5) * 8
sc = ax.scatter(
sub['x'],
sub['y'],
c=sub['component_id'],
s=sizes,
cmap='tab20',
alpha=0.85,
linewidth=0,
)
ax.set_title(epitope)
ax.set_xlabel('Layout X')
ax.set_ylabel('Layout Y')
ax.set_aspect('equal', adjustable='datalim')
fig.suptitle('CDR3 graph components by epitope', fontsize=12)
plt.tight_layout()
plt.show()